
伴t(11;17)(q23;q21)易位的急性早幼粒细胞白血病1例并文献复习
2013-09-12 11:12:50常晓丽董征刘莎姚波满秋红刘铁强李玉芳刘志强刘广贤艾辉胜郭梅
解放军医学杂志 2013年5期
常晓丽,董征,刘莎,姚波,满秋红,刘铁强,李玉芳,刘志强,刘广贤,艾辉胜,郭梅
伴t(11;17)(q23;q21)易位的急性早幼粒细胞白血病1例并文献复习
常晓丽,董征,刘莎,姚波,满秋红,刘铁强,李玉芳,刘志强,刘广贤,艾辉胜,郭梅
目的探讨伴t(11;17)(q23;q21)易位的急性早幼粒细胞白血病(APL)的临床特点、治疗及预后情况。方法报告1例APL伴t(11;17) (q23;21),分析其细胞形态学、免疫学、细胞遗传学及分子遗传学特点,并对近20年国内外相关文献进行复习。结果本例患者为男性,35岁,白细胞计数38.17×109/L,骨髓早幼粒细胞88.5%,细胞核规则,无Auer's小体,PLZF-RARA融合基因阳性。染色体核型:46,XY,t(11;17)(q23;q21)。诊断:APL伴t(11;17)(q23;q21)。治疗采用亚砷酸、全反式维甲酸(ATRA)和大剂量阿糖胞苷(2g/m2,总量24g,3个疗程)联合化疗,随访至2013年2月,患者完全缓解已10个月。国内外文献已报道伴t(11;17)(q23;q21)易位的APL患者20例,年龄23~75(48.9±16.3)岁,其中男性占90%。3例单用维甲酸治疗的患者中2例死于早期并发症,应用维甲酸联合诱导化疗的10例患者中6例在11~56个月死于复发,1例死于早期脓毒血症,提示大多数病例对维甲酸治疗疗效欠佳。结论伴t(11;17)(q23;q21)易位的APL是一种少见变异型,具有重要的形态学和临床特点。延长亚砷酸治疗时间,采用亚砷酸、全反式维甲酸和大剂量阿糖胞苷联合化疗方案可能对缓解病情和患者存活有益。
白血病,前髓细胞性,急性;易位,遗传;抗肿瘤联合化疗方案
急性早幼粒细胞白血病(acute promyelocytic leukemia,APL)的典型特征是伴有t(15;17)(q22;21)染色体易位,并形成特征性PML/RARA融合基因,其多数对维甲酸及砷剂治疗敏感[1-4]。但有5%~10%的APL患者缺乏这种典型的染色体改变[2-6],伴t(11;17)(q23;q21)易位的APL就是其中一种罕见而独特的变异类型,其基因学特点是11号染色体上的早幼粒细胞白血病锌指基因(promyelocytic leukemia zinc finger,PLZF)与位于17号染色体的RARA基因发生融合,而且此类患者对全反式维甲酸(ATRA)不敏感,对砷剂及化疗的反应目前亦无统一的报道[7-12]。军事医学科学院附属医院血液科经治1例伴t(11;17)(q23;q21)易位的APL患者,经亚砷酸、维甲酸联合化疗治疗取得较好疗效,已达持续完全缓解(complete response,CR)期,现就其临床和实验室特点并结合文献讨论报道如下。
1 病例资料
患者,男,35岁,因“周身乏力1年,发热16d”于2012-02-14入院。血常规:白细胞38.17×109/L,血红蛋白72g/L,血小板100×109/ L;出凝血功能:凝血酶原时间(PT) 15.2s,活动度57%,纤维蛋白原定量(FIB)0.47g/L,部分凝血活酶时间(APTT)不凝,凝血酶时间(TT) 21.3s,纤维蛋白降解产物(FDP)72.8μg/ml,伴全身多发出血倾向。骨髓形态学检查提示:①骨髓异常颗粒增多的早幼粒细胞占88.5%,胞核多规则,染色质固缩;②胞体大,胞质丰富,胞质粗颗粒型占31%,细颗粒型57.5%;③未见Auer's小体(图1A)。外周血白细胞分类:早幼粒细胞64%。免疫组化:过氧化物酶(POX)、特异性酯酶(CE)染色阳性率均为100%,非特异性酯酶(NSE)染色阳性率为68%,氟化钠(NaF)抑制试验阴性;PLZF-RARA融合基因定性阳性;PLZF-RARA/ABL1=86.78%。FISH分析间期细胞400个,未见含典型PML/RARA融合信号的细胞,而3R2G信号模式(无BCR/ABL融合基因的异常信号模式)细胞364个,占91%;2R1G1Y信号模式(DC-DFFISH融合信号特征)细胞24个,占6%,提示可能是涉及17q21的RARA基因的少见变异型易位(图1C)。染色体R显带核型分析示46,XY,t(11;17)(q23;q21) [3]/46,XY,?del(9q),t(11;17)[2]/46,XY[9]核型(图2A)。免疫分型:cMPO 77.7%、CD33 38.8%、CD13 92.8%、CD64 22.6%、CD15 26.1%、CD117 44.8%、CD56 63.4%。诊断为伴t(11;17)(q23;q21)易位的APL。
患者确诊当天(2012-02-18)起给予亚砷酸(10mg,1次/d,共55d)诱导化疗,亚砷酸治疗诱导第6天白细胞36.4×109/L,故辅以MA方案(米托蒽醌10mg,d1-3+阿糖胞苷200mg,d1-7),亚砷酸化疗3周复查骨髓异常早幼粒细胞占74.5%,提示未缓解。为此,化疗第24天加用维甲酸60mg,1次/d口服诱导分化治疗。第28天白细胞再次升至35.07×109/L,遂加用HA方案(高三尖杉酯碱3mg,d1-7,阿糖胞苷200mg,d1-7)再诱导化疗。亚砷酸共用55d,维甲酸共36d。期间患者外周血白细胞、骨髓比例变化见图3。停用亚砷酸后20d复查见骨髓中原始+早幼粒细胞占3.5%,外周血未见原始及早幼粒细胞,PLZF-RARA/ABL1=2.58%,FISH分析未见17q21的RARA基因变异(图1D),染色体R显带核型分析示46,XY(图2B),提示达形态学及遗传学完全缓解。
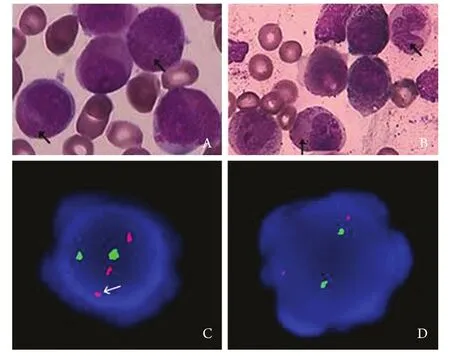
图1 诱导化疗前后骨髓形态学及FISH分析结果对比Fig.1 Comparison of marrow morphology and FISH analysis before and after induction chemotherapyA. Morphology of bone marrow before induction chemotherapy (Arrows indicate the nuclear in regular shape, cytoplasmic particles and no Auer's bodies in cytoplasm) (Wright's ×100); B. Morphology of bone marrow after induction chemotherapy (Arrows indicate the increased caryolobism and decreased cytoplasmic granules)(Wright's ×100); C. Abnormality of chromosome with 17q21 revealed by FISH analysis before induction chemotherapy (Arrow indicates fracture of chromosome 17 after labeled by PML-RARA probe); D. No abnormality was found of RARA gene with 17q21 by FISH analysis in CR patients after induction chemotherapy
2012-05-13至2012-11-27共给予大剂量阿糖胞苷(阿糖胞苷2g/m2,总量24g)3个疗程,以进行缓解后治疗。2013年1月复查PLZF-RARA/ABL1=0,提示获分子生物学完全缓解。随访至2013年2月,患者仍处于CR1期,无复发生存10个月。
2 文献复习
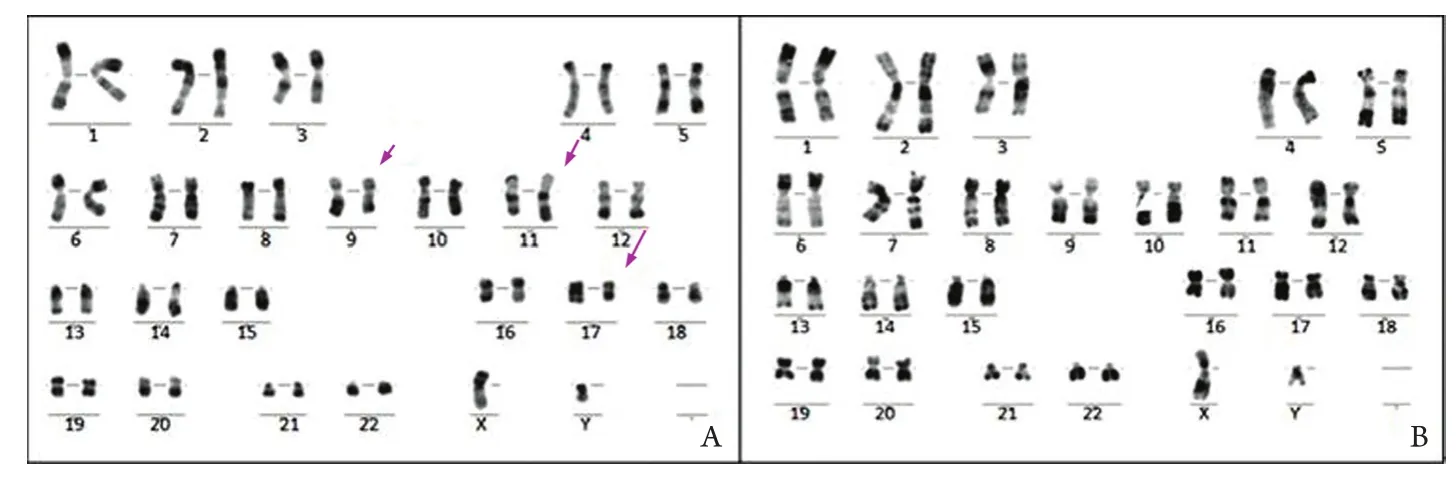
图2 诱导化疗前后染色体R显带核型分析结果Fig.2 Karyotype analysis of chromosomal R-banding before and after induction chemotherapyA. Chromosome abnormality with t(11;17)(q23;q21) and ?Del(9q) before induction chemotherapy (Arrows indicate the translocation of 11q23 and 17q21 and the doubtful deletion of 9q, respectively); B. Normal karyotype of CR patients after induction chemotherapy
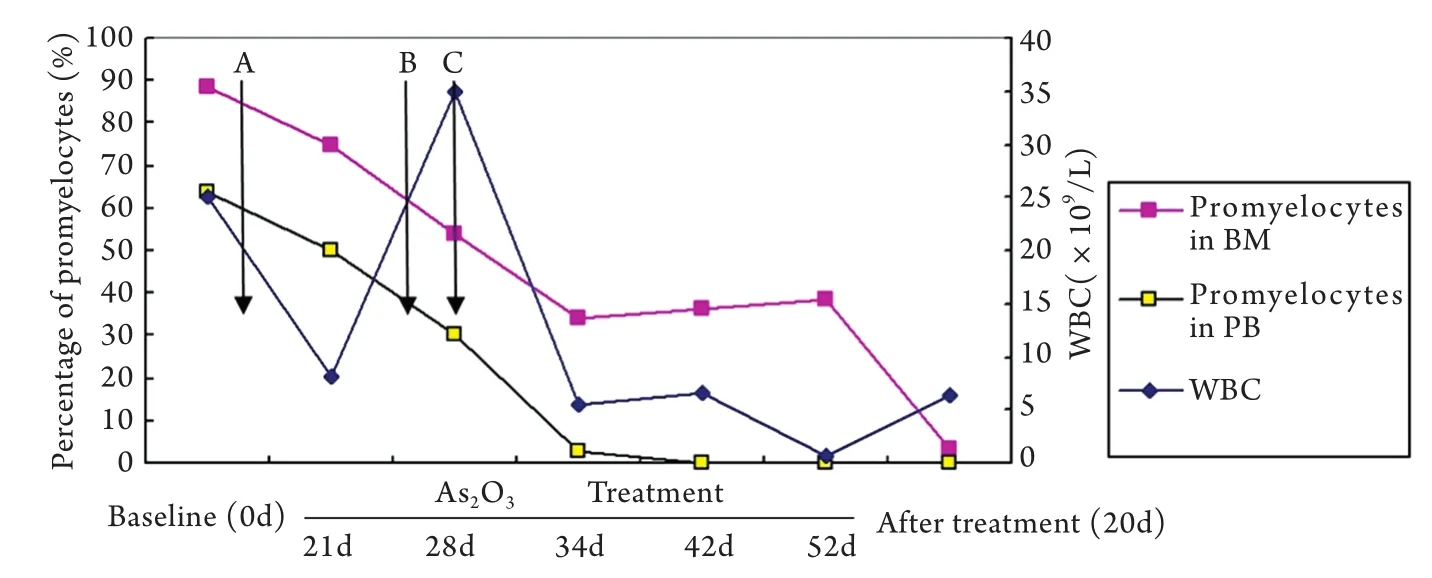
图3 诱导化疗期间骨髓、外周血早幼粒细胞比例及白细胞数变化Fig. 3 Changes of promyelocytes proportion in bone marrow (BM) and peripheral blood (PB) and counts of white blood cell (WBC) during induction chemotherapyMitoxantrone and Ara-C (MA) was added on the 6th day during the course of As2O3treatment; All Trans retinoic acid (ATRA) was added on the 24th day during the course of As2O3treatment; Homoharringtonine and Ara-C(HA) was added on the 28th day during the course of As2O3treatment
目前已报道的20例伴t(11;17)(q23;q21)易位的APL患者(表1)中,年龄分布在23~75岁(48.9±16.3岁),其中男性占90%,提示中老年男性可能为好发人群。所有患者均伴PLZF-RARA融合基因异常。20例患者中弥散性血管内凝血(DIC)的发生率达60%,与传统APL患者相当。既往报道的文献中,3例患者单用维甲酸治疗,其中2例死于早期并发症,10例采用维甲酸联合诱导化疗,其中6例在11~56个月死于复发,1例死于早期脓毒血症[13-24],提示大多数病例对维甲酸治疗效果欠佳。
3 讨 论
自上海报道世界首例伴t(11;17)(q23;q21)易位的APL以来[6],国外陆续报道并证实该型染色体易位的APL已接近20例。APL的特征性改变是产生了X/RARA融合基因,目前已报道与RARA基因融合的有PML、PLZF、NuMA、NPM1、FIP1L1、PRKAR1A、STAT5b及BCOR,其中以PML最多见,占90%~95%。而由t(11;17)(q23;q21)易位所形成的PLZF/RARA融合基因则是其中一种较为罕见的变异类型,占APL的0.80%~1.08%[24-25]。文献报道,PLZF/RARA与RARA/PLZF融合蛋白几乎同时表达于t(11;17)(q23;q21)APL患者中,RARA/PLZF作为一种癌基因,会干扰正常造血,可通过不同机制与PLZF/RARA共同作用导致t(11;17)(q23;q21)APL的患者发病,而且对ATRA及传统化疗耐药[6,26]。PLZF/RARA能阻断髓系分化中起重要作用基因的表达,从而使髓系细胞停滞在早期阶段,尤其是早幼粒细胞阶段,而RARA/PLZF可激活细胞周期关键调控因子如cyclinA2等,使停滞分化的早幼粒细胞生长加速,且不受正常调控,从而导致对ATRA耐药t(11;17)(q23;q21)APL的产生[6]。
不同于传统的PML/RARA重排,伴PLZF/ RARA基因重排的APL多表现为以下形态学特征:①细胞核规则;Pelger–Huët细胞增多,染色质固缩;②胞体大,胞质丰富,胞质内多为细颗粒;③多无Auer's小体[2,24,27]。Sainty等[2]的研究表明,规则核的出现是伴t(11;17)(q23;q21)APL的关键形态学特征,且这种形态学表现可直接受到RARA/PLZF融合蛋白的影响,提示RARA/PLZF与PLZF/RARA基因一样在该型APL的形成中不可或缺。另外,免疫表型方面也有一定特征有助于与传统APL进行鉴别,即CD56高表达[24]。有文献报道,CD56的高表达表明,与t(15;17)APL相比,t(11;17)APL可能来源于起源更早的白血病前体细胞[2]。
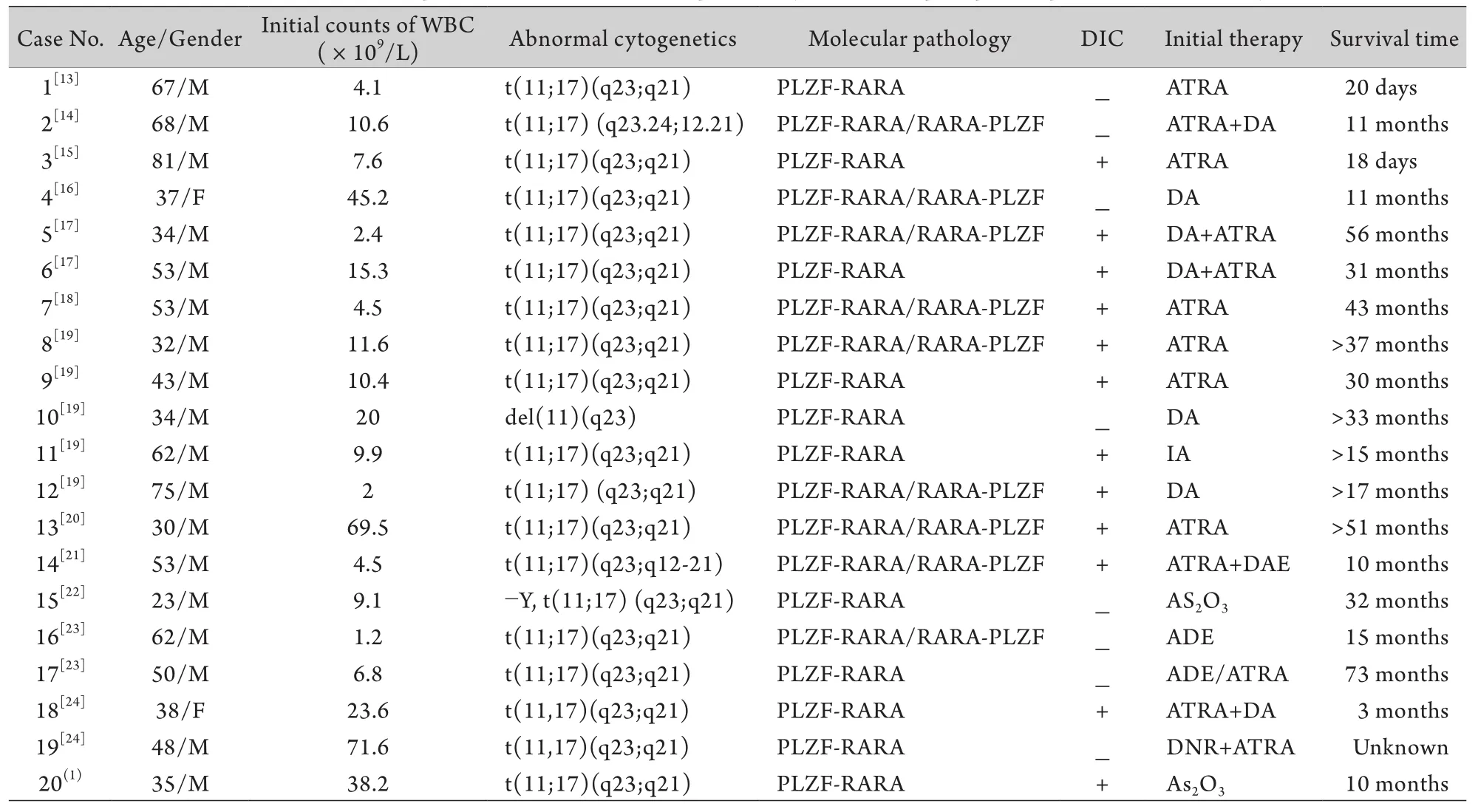
表1 国内外已报道的20例伴t(11;17)(q23;q21)APL患者的临床特征Tab. 1 Clinical features of 20 patients with APL accompanied by t(11;17)(q23;q21) reported domestically and aboard
目前,国内外对伴有t(11;17)(q23;q21)易位的APL的治疗尚无统一的方案,但大多数文献报道,其早期缓解率较低、易复发,治疗效果不佳,对单纯ATRA或亚砷酸治疗不敏感,联合化疗的疗效尚不明确;文献[13-24]报道,3例此类患者单用维甲酸治疗,其中2例死于早期并发症,10例采用维甲酸联合诱导化疗,其中6例在11~56个月死于复发,1例死于早期脓毒血症,提示大多数病例对维甲酸治疗疗效欠佳。但Petti等[8]曾报道过3例单用ATRA或联合G-CSF治疗达到完全缓解的病例。分析原因如下:该患者白血病细胞未表达RARA/PLZF融合蛋白,而后者被认为在APL形成及ATRA耐药机制中发挥重要作用;延长的ATRA治疗可能有助于取得更好疗效。尚有Kitamura等[11]报道2例通过ATRA联合G-CSF治疗达到完全缓解的病例。
本研究报道1例APL患者伴t(11;17)(q23;q21),发病早期合并DIC,其形态学特点符合前述典型特征的改变,并可见CD56高表达,经以亚砷酸为基础的联合化疗取得较好疗效。分析原因如下:本例在诱导治疗中延长了亚砷酸的疗程,并同时联合化疗和ATRA治疗,提高了完全缓解的可能性;本例治疗过程中采用了大剂量阿糖胞苷作为完全缓解后治疗,该举措可能对杀灭残留白血病细胞、维持缓解状态有重要作用。
综上,伴t(11;17)(q23;q21)易位的APL是一种罕见的变异型,具有重要的形态学、免疫学和临床特点,延长亚砷酸治疗时间、联合化疗及ATRA治疗有可能对其早期缓解及较长的无病生存有益。
[1] Lallemand-Breitenbach V, Zhu J, Chen Z, et al. Curing APL through PML/RARA degradation by As2O3[J]. Trends Mol Med, 2012, 18(1): 36-42.
[2] Sainty D, Liso V, Cantu-Rajnoldi A, et al. A new morphologic classification system for acute promyelocytic leukemia distinguishes cases with underlying PLZF/RARA gene rearrangements [J]. Blood, 2000, 96(4): 1287-1296.
[3] Zuo HL, Peng EL, Liu TQ, et al. Reproduction and appraisal of an animal model of acute myelomonocytic leukemia in the CB6F1generation mice[J]. Med J Chin PLA, 2011, 36(1): 53-57. [左洪莉, 彭恩兰, 刘铁强, 等. CB6F1小鼠急性粒单核细胞白血病M4(AML-M4)模型的建立及鉴定[J]. 解放军医学杂志, 2011, 36(1): 53-57.]
[4] 王荷花, 欧阳涓, 许多荣, 等. 急性早幼粒细胞白血病分化综合征临床特征及对预后的影响研究[J]. 中国实用内科杂志, 2012, 32(2): 132-134.[Wang HH, Ouyang J, Xu DR, et al. Differentiation syndrome in patients with acute promyelocytic leukemia:clinical features and prognosis[J]. Chin J Pract Intern Med, 2012, 32(2): 132-134.]
[5] Chen Z, Guidez F, Rousselot P, et al. PLZF-RAR alpha fusionproteins generated from the variant t(11;17)(q23;q21) translocation in acute promyelocytic leukemia inhibit ligand-dependent transactivation of wild-type retinoic acid receptors[J]. Proc Natl Acad Sci USA, 1994, 91(3): 1178-1182.
[6] Cao WJ. Role of RARα-PLZF in the developmeent of APL with t (11; 17)(q23; q21)[J]. Foreign Med Sci(Section Blood Transfus Heamatol), 2001, 24(3): 197-199. [曹文俊. RARα-PLZF在t(11; 17)(q23; q21)APL发病中的作用[J]. 国外医学输血及血液学分册, 2001, 24(3): 197-199.]
[7] Park DJ, Vuong PT, De Vos S, et al. Comparative analysis of genes regulated by PML/RAR alpha and PLZF/RAR alpha in response to retinoic acid using oligonucleotide arrays[J]. Blood, 2003, 102(10): 3727-3736.
[8] Petti MC, Fazi F, Gentile M, et al. Complete remission through blast cell differentiation in PLZF/RARalpha-positive acute promyelocytic leukemia: in vitro and in vivo studies[J]. Blood, 2002, 100(3): 1065-1067.
[9] Rice KL, Hormaeche I, Doulatov S, et al. Comprehensive genomic screens identify a role for PLZF-RARalpha as a positive regulator of cell proliferation via direct regulation of c-MYC[J]. Blood, 2009, 114(27): 5499-5511.
[10] Boukarabila H, Saurin AJ, Batsche E, et al. The PRC1 Polycomb group complex interacts with PLZF/RARA to mediate leukemic transformation[J]. Genes Dev, 2009, 23(10): 1195-1206.
[11] Kitamura K, Hoshi S, Koike M, et al. Histone deacetylase inhibitor but not arsenic trioxide differentiates acute promyelocytic leukaemia cells with t(11;17) in combination with all-trans retinoic acid[J]. Br J Haematol, 2000, 108(4): 696-702.
[12] Spicuglia S, Vincent-Fabert C, Benoukraf T, et al. Characterisation of genome-wide PLZF/RARA target genes[J]. PLoS One, 2011, 6(9): e24176.
[13] Chen SJ, Zelent A, Tong JH, et al. Rearrangements of the retinoic acid receptor alpha and promyelocytic leukemia zinc finger genes resulting from t(11;17)(q23;q21) in a patient with acute promyelocytic leukemia[J]. J Clin Invest, 1993, 91(5): 2260-2267.
[14] Guidez F, Huang W, Tong JH, et al. Poor response to all-trans retinoic acid therapy in a t(11;17) PLZF/RAR alpha patient[J]. Leukemia, 1994, 8(2): 312-317.
[15] Frankel SR, Eardley A, Lauwers G, et al. The "retinoic acid syndrome" in acute promyelocytic leukemia[J]. Ann Intern Med, 1992, 117(4): 292-296.
[16] Scott AA, Head DR, Kopecky KJ, et al. HLA-DR-, CD33+, CD56+, CD16- myeloid/natural killer cell acute leukemia: a previously unrecognized form of acute leukemia potentially misdiagnosed as French-American-British acute myeloid leukemia-M3[J]. Blood, 1994, 84(1): 244-255.
[17] Licht JD, Chomienne C, Goy A, et al. Clinical and molecular characterization of a rare syndrome of acute promyelocytic leukemia associated with translocation (11;17)[J]. Blood, 1995, 85(4): 1083-1094.
[18] Grimwade D, Gorman P, Duprez E, et al. Characterization of cryptic rearrangements and variant translocations in acute promyelocytic leukemia[J]. Blood, 1997, 90(12): 4876-4885.
[19] Grimwade D, Biondi A, Mozziconacci MJ, et al. Characterization of acute promyelocytic leukemia cases lacking the classic t(15;17): results of the European Working Party. Groupe Francais de Cytogenetique Hematologique, Groupe de Francais d'Hematologie Cellulaire, UK Cancer Cytogenetics Group and BIOMED 1 European Community-Concerted Action "Molecular Cytogenetic Diagnosis in Haematological Malignancies"[J]. Blood, 2000, 96(4): 1297-1308.
[20] Jansen JH, De Ridder MC, Geertsma WM, et al. Complete remission of t(11;17) positive acute promyelocytic leukemia induced by all-trans retinoic acid and granulocyte colonystimulating factor[J]. Blood, 1999, 94(1): 39-45.
[21] Culligan DJ, Stevenson D, Chee YL, et al. Acute promyelocytic leukaemia with t(11;17)(q23;q12-21) and a good initial response to prolonged ATRA and combination chemotherapy[J]. Br J Haematol, 1998, 100(2): 328-330.
[22] George B, Poonkuzhali B, Srivastava VM, et al. Hematological and molecular remission with combination chemotherapy in a patient with PLZF-RARalpha acute promyelocytic leukemia (APML)[J]. Ann Hematol, 2005, 84(6): 406-408.
[23] Jovanovic JV, Rennie K, Culligan D, et al. Development of realtime quantitative polymerase chain reaction assays to track treatment response in retinoid resistant acute promyelocytic leukemia [J]. Front Oncol, 2011, 1: 35.
[24] Rohr SS, Pelloso LA, Borgo A, et al. Acute promyelocytic leukemia associated with the PLZF-RARA fusion gene: two additional cases with clinical and laboratorial peculiar presentations[J]. Med Oncol, 2012, 29(4): 2345-2347.
[25] Jacomo RH, Melo RA, Souto FR, et al. Clinical features and outcomes of 134 Brazilians with acute promyelocytic leukemia who received ATRA and anthracyclines[J]. Haematologica, 2007, 92(10): 1431-1432.
[26] Guidez F, Parks S, Wong H, et al. RARalpha-PLZF overcomes PLZF-mediated repression of CRABPI, contributing to retinoid resistance in t(11;17) acute promyelocytic leukemia[J]. Proc Natl Acad Sci U S A, 2007, 104(47): 18694-18699.
[27] Wakim JJ, Tirado CA. Acute promyelocytic leukemia lacking the classic translocation t(15; 17)[M]//Koschmieder S. Myeloid leukemia -- clinical diagnosis and treatment. Rijeka: InTech, 2012. 12-228.
Acute promyelocytic leukaemia with translocation of t(11;17)(q23;q21): a case report and review of literature
CHANG Xiao-li1, DONG Zheng2, LIU Sha2, YAO Bo2, MAN Qiu-hong2, LIU Tie-qiang2, LI Yu-fang2, LIU Zhi-qiang2, LIU Guang-xian2, AI Hui-sheng2, GUO Mei2*
1Postgraduate Medical School of PLA, Beijing 100853, China
2Department of Hematology, Affiliated Hospital, Academy of Military Medical Sciences, Beijing 100071, China
*
, E-mail: guom196801@yahoo.com.cn
ObjectiveTo study the clinical attributes, treatment and prognosis of acute promyelocytic leukaemia (APL) with translocation of t(11;17)(q23;q21).MethodsA case of APL with t(11;17)(q23;q21) was reported, and the cytomorphology, immunology, cytogenetics and molecular genetics of the patient were analyzed. The related literature published in recent 20 years domestically and abroad was reviewed.ResultsThe case herewith studied was a male patient aged 35 years with leucocyte count (WBC) of 38.17×109in peripheral blood, and 88.5% of promyelocytes in bone marrow. Marrow karyotype analysis showed 46, XY, t(11;17)(q23;q21) and PLZF-RARA rearrangement was detected. The patient was definitely diagnosed as APL with t(11;17) (q23;q21). Complete morphological and genetic remission was achieved after a combined chemotherapy of As2O3and ATRA, then a molecular biological remission was achieved after the treatment of 3 courses of large dose cytarabine. The patient was followed up to February 2013 with disease-free survival of nearly 10 months. Up to date, 20 cases of APL with t(11;17)(q23;q21) have been reported domestically and abroad, and their clinical attributes were reviewed. For the 20 cases, the mean age was 48.9±16.3 years and the male/female ratio was 9:1. Among the patients, males aged over 45 years accounted for 55%, implying that the elderly males may be high-risk population. The incidence of DIC was 60%, most of which got poor curative effect of ATRA.ConclusionAPL with t(11;17)(q23;q21) is a very rare illness with distinct morphological changes and clinical characteristics. Prolonged combined chemotherapy with As2O3and ATRA with large dose cytarabine may be beneficial to attain a remission and prolong survival.
leukemia, promyelocytic, acute; translocation, genetic; antineoplastic combined chemotherapy protocols
R733.712
A
0577-7402(2013)05-0378-05
2013-03-12;
2013-03-20)
(责任编辑:沈宁)
国家自然科学基金(30200116)
常晓丽,住院医师,硕士研究生。主要从事恶性血液病方面的研究
100853 北京 解放军医学院[常晓丽(现在军事医学科学院附属医院血液科)];100071 北京 军事医学科学院附属医院血液科(董征、刘莎、姚波、满秋红、刘铁强、李玉芳、刘志强、刘广贤、艾辉胜、郭梅)
郭梅,E-mail:guom196801@yahoo.com.cn
This work was supported by the National Natural Science Foundation of China(30200116)
猜你喜欢
中国卫生标准管理(2022年21期)2023-01-03 02:36:34
生物技术进展(2019年3期)2019-05-28 03:46:40
中国继续医学教育(2015年2期)2016-01-06 01:36:16
医学研究杂志(2015年2期)2015-06-10 06:45:00
家庭医学(2014年8期)2014-09-12 13:41:42
振动、测试与诊断(2014年6期)2014-03-01 01:14:50
现代检验医学杂志(2014年1期)2014-02-06 01:29:31
现代检验医学杂志(2014年5期)2014-02-02 02:51:35
中国实用医药(2013年9期)2013-02-02 13:59:19
中国医学科学院学报(2012年3期)2012-03-25 13:58:49
