
金刚烷-1-甲酸衍生物的合成*
2013-09-01 02:12:36韦雪琴吴彩婷李子成
合成化学 2013年6期
邓 磊,谭 锐,韦雪琴,唐 杰,吴彩婷,李子成
(四川大学 化工学院,四川 成都 610065)
金刚烷衍生物具有重要的生物活性[1,2]。根据生物电子等排体原理,氟具有氢类似的原子半径,将金刚烷衍生物中的叔碳氟代后可能会提高其生物活性或降低其毒副作用。
通过直接氟代法或用其他基团取代后再氟代可以在金刚烷甲酸的三个叔碳原子上分别或同时引入一至三个氟原子。但是这些方法都有缺陷,例如,电化学方法需要特殊设备,而且无法大规模生产[3]。金刚烷-1-甲酸乙酯用BrF3为氟化剂可同时在3个叔碳原子上引入氟,但需用氟气[4,5]。此外,采用二乙胺基三氟化硫(DAST)[6,7]或四氟化硫取代羟基进行氟代,且收率高[8],但DAST或四氟化硫价格昂贵,腐蚀性强,反应条件苛刻并且不易操作。而五氟化碘只能在金刚烷的叔碳原子上选择性的引入一个或两个氟原子[9],也要使用氟气。以上这些氟代试剂都难以实现工业化生产。
吡啶聚氟化氢(PPHF,70%HF吡啶溶液,也称Olah试剂)的沸点为80℃,可以进行回收利用,其腐蚀性小,价格便宜,而且反应条件温和,在氟代反应中有着广泛的应用[10]。单独使用PPHF可以直接取代烃类化合物仲或叔碳上的羟基。采用PPHF对溴或氯进行取代反应时,需要和四氟硼酸硝酰金翁(NO2+BF4-)一起使用[11,12]。此外,二甲醚/聚氟化氢也可取代仲或叔碳原子上的羟基进行氟代反应[13]。
由于金刚烷-1-甲酸(1)及其取代衍生物可通过酰胺重排得到1-金刚烷胺,而且1性质稳定。为此,本文以1为起始原料,PPHF为氟化剂,经取代反应制得 3-氟金刚烷-1-甲酸(3),收率83%。以高锰酸钾为氧化剂,在碱性条件3被氧化成5-羟基-3-氟金刚烷-1-甲酸(4);将其羟基转化为甲烷磺酸酯后再用PPHF取代合成了3,5-二氟金刚烷-1-甲酸(6,Scheme 1),收率70%。其结构经1H NMR,MS和元素分析确证。
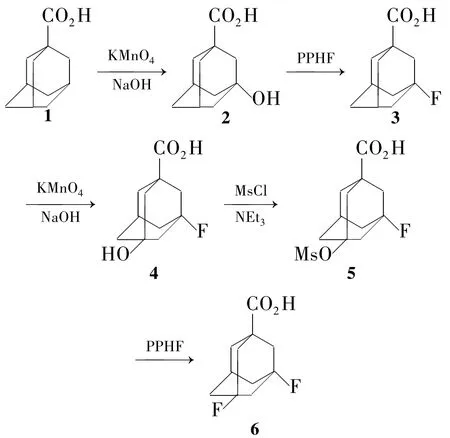
Scheme 1
1 实验部分
1.1 仪器与试剂
XRC-1型显微熔点仪(温度未校正);Varian INOVA 400(400 MHz)型核磁共振仪(CDCl3为溶剂,TMS为内标);BRUKER Amazon SL型质谱仪;Perkin Elmer 2400型元素分析仪。
3-羟基金刚烷-1-甲酸(2)和 4 参考文献[6,14]方法合成;PPHF,南京阿托化工有限公司,其余所用试剂均为分析纯。
1.2 合成
(1)3的合成
在500 mL聚四氟乙烯反应瓶中加入2 29 g(148 mmol)和 PPHF 200 mL,密闭,搅拌下于50℃反应10 h(TLC监测)。自然冷却至室温,缓慢倾入600 mL冰水中,过滤,滤饼用水洗涤,真空干燥得白色固体3 24 g,收率83%,m.p.153℃~154 ℃(154 ℃~156 ℃[8]);1H NMR δ:1.61(s,2H),1.82(s,4H),1.88(s,4H),2.03(d,J=5.2 Hz,2H),2.36(s,2H);ESI-MS m/z:197.1(100%){[M-H]-};Anal.calcd for C11H15O2F:C 66.65,H 7.63;found C 66.73,H 7.58。
(2)6的合成
在反应瓶中加入4 7.8 g(36.5 mmol)和二氯甲烷50 mL,搅拌使其溶解;加入三乙胺15 mL,冰浴冷却,缓慢滴加甲磺酰氯8.7 g(76 mmol),滴毕,于室温反应3 h。加入2 mol·L-1盐酸50 mL,分液,水相用二氯甲烷(2×30 mL)萃取,合并有机相,用饱和食盐水(2×30 mL)洗涤,无水硫酸镁干燥,回收溶剂至干得淡黄色固体5 8.5 g,未经纯化直接投入下步反应。
在100 mL聚四氟乙烯反应瓶中加入5和PPHF 50 mL,密闭,搅拌下于50℃反应3 h(TLC监测)。冷却至室温,倾入100 mL冰水中,析出固体,过滤,滤饼用水洗涤,干燥得白色固体6 3.2 g。滤液用二氯甲烷(2×30 mL)萃取,合并有机相,用饱和食盐水(2×20 mL)洗涤,无水硫酸镁干燥,回收二氯甲烷至干,残余物用混合溶剂[V(丙酮)∶V(水)=8 ∶2]重结晶得白色固体6 2.3 g,合并两次所得6,收率70%,m.p.161 ℃~163℃(162 ℃~ 164 ℃[6]);1H NMR δ:1.75(brs,2H),1.85(brs,4H),2.04(brs,4H),2.11(m,2H),2.55(brs,1H);ESI-MS m/z:215.1(100%){[M-H]-};Anal.calcd for C11H14O2F2:C 61.10,H 6.53;found C 61.38,H,6.54。
2 结果与讨论
在氟代反应时,我们曾尝试采用NEt3·3HF为氟代剂,但实验结果显示NEt3·3HF均不与2和5反应,可能是NEt3·3HF的氟代能力较差。先将2转化成3-甲磺酰氧基金刚烷-1-甲酸后再使用NaF和NEt3·3HF进行氟代,也未见反应发生。但2与PPHF(7 mL·g-1)在50℃下反应10 h后,TLC监测显示反应已进行完全。将反应液倒入冰水中,直接析出固体,3的收率83%。
当采用与2相同的氟代方法对4进行氟代时,则不发生反应。可能是3-位氟的存在除低了金刚烷环上其它羟基的反应活性所致。将4转化成活性更高的甲磺酸酯5,未经纯化直接进行氟代,TLC监测显示3 h后反应即已完成。鉴于金刚烷的甲磺酸酯更易氟代,将2也转化成了它的甲磺酸酯,结果表明反应只需1.5 h即可完成,但收率只有78%。
实验中我们试图将7或8用PPHF氟代成9(Chart 1),但未获成功,原因除了与PPHF反应活性低有关外,还可能与空间位阻及氟原子的强吸电子效应有关。
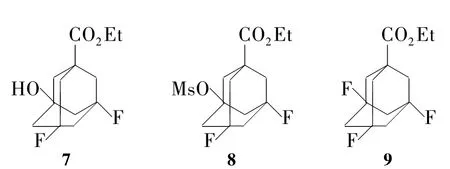
Chart 1
3 结论
采用PPHF对金刚烷衍生物的羟基或甲磺酸基进行氟代,成功合成了3-氟金刚烷-1-甲酸和3,5-二氟金刚烷-1-甲酸。将羟基转化成甲磺酸酯后提高了反应活性,同时大大缩短氟代时间。
[1] Spasov A A,Khamidova T V,Bugaeva L I,et al.Adamantane derivatives:Pharmacological and toxicological properties[J].Pharm Chem J,2000,34(1):1-7.
[2] Geldenhuys W J,Malan S F,Bloomquist J R,et al.Pharmacology and structure-activity relationships of bioactive polycyclic cage compounds:A focus on pentacycloundecane derivatives[J].Med Res Rev,2005,25(1):21-48.
[3] Aoyama M,Fukuhara T,Hara S.Selective fluorination of adamantanes by an electrochemical method[J].J Org Chem,2008,73(11):4186-4189.
[4] Shishimi T,Hara S.Selective introduction of fluorine atoms to the tert-carbons of functionalized adamantanes by BrF3[J].J Fluorine Chem,2013,145:128-131.
[5] Rozen S,Rechavi D,Hagooly A.Novel reactions with the underutilized BF3-The chemistry with nitrile[J].J Fluorine Chem,2001,111(2):161-165.
[6] Jasys V J,Lombardo F,Appleton T A,et al.Preparation of fluoroadamantane acids and amines:Impact of bridgehead fluorine substitution on the solution-and solid-state properties of functionalized adamantanes[J].J Am Chem Soc,2000,122(3):466-473.
[7] Middleton W J.New fluorinating reagents.Dialkylaminosulfur fluorides[J].J Org Chem,1975,40(5):574-578.
[8] Adcock W,Kok G B.Transmission of polar substituent effects in saturated systems:Synthesis and19F NMR study of 3-substituted adamant-1-yl fluorides[J].J Org Chem,1987,52:356-364.
[9] Hara S,Aoyama M.Direct fluorination of adamantanes with iodine pentafluoride[J].Synthwsis-Stuttgart,2008,(16):2510-2512.
[10] Olah G A,Welch J T,Vankar Y D,et al.Synthetic methods and reactions.63.Pyridinium poly(hydrogen fluoride)(30% pyridine-70% hydrogen fluoride):A convenient reagent for organic fluorination reactions[J].J Org Chem,1979,44(22):3872-3881.
[11] Olah G A,Shih J G,Krishnamurthy V V,et al.Preparation and13C NMR spectroscopic study of fluoroadamantanes and fluorodiamantanes:Study of13C-19F NMR coupling constants[J].J am Chem Soc,1984,106(16):4492-4500.
[12] Olah G A,Shih J G,Singh B P,et al.Ionic fluorination of adamantane,diamantane,and triphenylmethane with NO+BF4-/pyridine polyhydrogen fluoride(PPHF)[J].J Org Chem,1983,48(19):3356-3358.
[13] Bucsi I,Torok B,Marco A I,et al.Stable dialkyl ether/poly(hydrogen fluoride)complexes:Dimethyl ether/poly(hydrogen fluoride),A new,convenient,and effective fluorinating agent[J].J Am Chem Soc,2002,124(26):7728-7736.
[14] Anderson G L,Burks W A,Harruna I I.Novel synthesis of 3-fluoro-1-aminoadamantane and some of its derivatives[J].Synthetic Commun,1988,18(16-17):1967-1974.
猜你喜欢
中国典型病例大全(2022年9期)2022-04-19 22:12:45
中国民间疗法(2021年17期)2021-11-04 08:39:48
——以塔里木盆地塔中地区凝析油为例
石油实验地质(2021年5期)2021-11-01 06:55:44
地球科学与环境学报(2020年2期)2020-03-26 10:01:28
石油与天然气地质(2018年4期)2018-08-01 06:36:56
中国卫生标准管理(2015年17期)2016-01-20 09:26:44
中国当代医药(2015年9期)2015-03-01 02:02:07
中国当代医药(2015年9期)2015-03-01 02:02:03
中华皮肤科杂志(2014年4期)2014-12-19 12:55:56
中国塑料(2014年12期)2014-10-17 02:49:41
