
自噬在缺血预适应减少急性心肌缺血-再灌注损伤中的作用*
2013-08-29 09:43:52刘艺徐卫娟柯丽李云桥彭雯
微循环学杂志 2013年3期
刘艺 徐卫娟 柯丽 李云桥 彭雯,#
缺血预适应(Ischemic Preconditioning,IPC)[1]是机体减少缺血再灌注(Ischemia-Reperfusion,I/R)损伤最有效的内源性保护措施之一。经IPC 干预I/R 心肌后,其白介素-1β(Interleukin-1β,IL-1β)、IL-6和肿瘤坏死因子-α(Tumor Nectosis Factor-α,TNF-α)含量减少,可能是其保护I/R 心肌的一个重要途径[2]。
自噬(Autophagy)广泛存在于真核细胞中,可以降解和循环利用胞内物质如聚集的蛋白、某些病原体(分支杆菌和疱疹病毒)和多余或者损坏的细胞器(线粒体)等;当缺氧或营养物质缺乏时,自噬可通过降解胞内物质产生氨基酸和脂肪酸为机体提供能量,并促进机体蛋白和细胞器的合成。越来越多的证据显示自噬的发生可减轻心肌I/R 损伤程度[3];研究[4]发现多种信号均可诱导自噬,如蛋白激酶C(Protein Kinase C,PKC)、活性氧簇(Reactive Oxygen Species,ROS)、一氧化氮(NO)和腺苷一磷酸激活蛋白激酶(Adenosine Monophosphate-activated Protein Kinase,AMPK)等。心肌微管相关蛋白1轻链3-II(LC3-II)是自噬的标记分子,定位于自噬前体和自噬体,可用于评价自噬强度。
本实验采用冠脉结扎术建立大鼠急性心肌I/R模型,研究IPC减少大鼠心肌I/R 损伤及自噬通路在其中的作用。
1 材料与方法
1.1 实验动物和分组
雄性SD 大鼠32只,体重200~250g。均分为假手术组(Sham 组)、I/R 组、IPC 组和自噬抑制剂渥曼青霉素组(Wort组)。
1.2 主要试剂
兔抗鼠LC3B抗体(抗LC3-II一抗,美国Sigma公司);BCA 法蛋白浓度测定试剂盒和Protein A+G Agarose(中国碧云天生物公司);原位末端脱氧核苷酸转移酶标记(Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick-end Labeling,TUNEL)试剂盒(DAB法,瑞士Roche公司)。
1.3 各组大鼠处理方法
1.3.1 大鼠急性心肌I/R 模型的制备:20%乌拉坦0.6ml/100g腹腔注射麻醉后,将大鼠仰卧固定于解剖台上,四肢皮下连接心电图电极,监测标准Ⅱ导联心电图。气管插管,连接小动物呼吸机(DH-140浙江)通气。呼吸机设置:潮气量8~10ml,呼吸频率60~64次/min,呼吸比1∶3。于胸骨左缘2~3肋间开胸,“T”形剪开心包,在左心耳下缘与肺动脉圆锥之间找到冠脉动脉左前降支,将6-0无损伤线用手术缝合针穿至冠脉下,进针深度控制在0.1cm,宽度0.1~0.2cm,之后对各组大鼠进行如下处理:(1)Sham 组只穿线,不结扎冠脉,观察150min;(2)I/R 组收紧结扎线并打结,心电图Ⅱ导联S-T 段呈弓背向上抬高,同时肉眼见结扎区变白,证实心肌缺血。30min 后松开结扎线,再灌注120min;(3)IPC组于I/R 前结扎冠脉5min、松开再灌注5min,重复3次后,再结扎30min,再灌注120min;(4)Wort组:腹腔注射Wort(15μg/Kg),15min后结扎冠脉5min,松开再灌注5min,重复3次后,再结扎30min,再灌注120min。Sham 组、I/R组和IPC组同时腹腔注射等体积生理盐水。
脱臼处死各组大鼠,迅速取出心脏,用冰冷生理盐水冲洗,剪去非左心室部分,再剪取白色梗死心肌。分成三部分,一部分用于检测心肌细胞凋亡率;另一部分用于制作超薄切片,电镜观察自噬体数量和线粒体超微结构变化;剩下部分-80℃冰箱中冷藏备检LC3-II蛋白表达。Sham 组剪取相同部位心肌,作同样处理。
1.4 检测指标和方法
1.4.1 心肌细胞凋亡检测:将各组心肌样本置于4%多聚甲醛中,4℃固定24h后,石蜡包埋制成组织切片。按TUNEL 试剂盒说明书进行染色操作。由2个不同观察者在每张切片上随机取5个不重复视野进行凋亡细胞计数,以凋亡细胞数占心肌细胞总数的百分比作为凋亡指数(Apoptotic Index,AI)。细胞核呈棕黄色或棕褐色者为凋亡细胞。
1.4.2 自噬体和线粒体超微结构观察:将各组心肌标本于4℃2.5%戊二醛固定液中预固定5min,待软组织稍变硬后再将组织修剪成截面积为1mm2的5mm 长条(2条),室温固定2h;用pH 7.4的PBS冲洗;l%锇酸固定lh 再冲洗,梯度乙醇脱水,环氧树脂(EPON812)包埋,超薄切片机(德国LEICA ULTRACUT UCT)切片;醋酸双氧铀及柠檬酸双重染色,透射电子显微镜(荷兰Tecnai G212型)观察心肌自噬体数量及线粒体超微结构。
1.4.3 Western Blotting检测LC3-II蛋白表达:从-80℃冰箱中取出各组心肌标本,加入3倍体积的组织裂解液,冰水浴中充分研磨成组织匀浆,放置10min;4℃15000g离心10min;加入蛋白上样液,沸水煮5min。BCA 法定量蛋白浓度。加等量2×SDS上样缓冲液,95℃变性5min。每孔加40μg胞浆蛋白,经12%SDS-PAGE分离,4℃下100V 恒压转膜,室温下用5%脱脂奶粉封膜2h,加入兔抗鼠抗LC3-II(1∶500)或内参GAPDH(1∶800)一抗,于4℃孵育过夜;洗膜后与辣根过氧化酶标记的羊抗兔IgG(1∶5000)室温孵育1h;漂洗后ECL发光,常规显影。用Gel pro 4.0凝胶光密度分析软件进行分析,结果以目的条带灰度/内参条带灰度比值表示相对表达量。
1.5 统计学处理
使用SPSS 17.0软件进行统计学分析,计量数据采用均数±标准差(±s)表示,多组均数间比较采用方差分析,两两比较采用Fisher LSD 法;均采用双侧检验;P<0.05为差异有统计学意义。
2 结果
2.1 各组心肌细胞凋亡情况
凋亡细胞以心肌细胞为主,也可见淋巴细胞和血管内皮细胞。Sham 组仅见极少数散在凋亡细胞;I/R 组凋亡细胞较多,AI较Sham 组明显增加(P<0.01);IPC组凋亡减少,AI较I/R 组明显降低(P<0.01);而Wort组凋亡细胞最多,AI较IPC组明显增加(P<0.01)。见图1和图2。

图2 各组心肌细胞AI比较
2.2 自噬体和线粒体超微结构
电镜下,Sham 组未见明显自噬体,线粒体和心肌细胞形态结构均正常;I/R 组可见少量自噬泡,线粒体明显肿胀,嵴突紊乱、断裂或消失,心肌细胞水肿明显,肌小节明暗带模糊不清,肌丝断裂溶解,细胞器减少、结构破坏,胞浆内形成大小不等的空腔和空泡,细胞膜破裂,胞内容物泄漏等;IPC 组与I/R组相比,自噬泡数量增多,心肌细胞超微结构损伤减轻;Wort组心肌细胞超微结构破坏较IPC 组严重,且未见明显自噬泡。见图3。
2.3 各组LC3-II蛋白表达
Sham 组心肌组织LC3-II蛋白表达较少;I/R组及IPC组表达明显增多(P<0.05);IPC组较I/R组上调更显著(P<0.05)。见图4。
[本文图1、图3见封2]
3 讨论
自噬作为有别于细胞凋亡的第二类程序性细胞死亡途径,已证实广泛存在于真核细胞内,是一种溶酶体依赖性降解途径。在机体细胞发育、蛋白合成、组织重塑及环境适应等方面发挥着极其重要的作用[5]。自噬最基本的功能是当细胞营养物质不足时通过降解胞内的蛋白质和细胞器,满足细胞必要的能量需求和合成反应,从而保护机体应对各种应激状况。
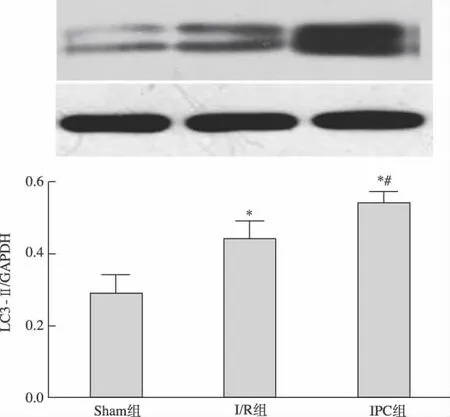
图4 各组大鼠心肌组织LC3-II蛋白表达
IPC是一种内源性心肌保护方法,既往研究证实IPC可减少心肌I/R 过程中过多的氧自由基,防止细胞内钙超载,降低能量消耗和抑制去甲肾上腺素的过度分泌等机制,发挥心肌保护作用[6]。
Decker等[7]最早报道了I/R 可诱导心肌细胞自噬的发生,该研究发现兔的心肌细胞在经历了20~40min的缺氧后,开始出现自噬泡,而当氧供恢复后,自噬泡数量明显增加,且在急性或慢性缺血条件下自噬均可被启动,在再灌注阶段自噬又会进一步增强;此外,Kim 等[8]观察到处于冬眠状态的心肌细胞内,也可观察到自噬增强,同时心肌细胞的凋亡水平下降,心肌梗死面积减少。可见自噬可通过降解胞内物质产生氨基酸和脂肪酸,为机体提供能量,促进机体蛋白质和细胞器的合成,从而维持缺血缺氧状态下细胞的存活。
本实验通过建立大鼠急性心肌I/R 模型,观察到I/R 组有少量自噬泡,心肌细胞线粒体肿胀,细胞器结构破坏明显;IPC组与I/R 组相比,自噬泡数量明显增多,细胞器如线粒体损伤也减轻;而Wort组心肌细胞超微结构破坏较IPC 组严重,且未见明显自噬泡。LC3是酵母自噬相关基因Atg 8的类似物,其前体(ProLC3)形成后,被Atg 4剪切形成胞浆可溶性形式的LC3-I,并暴露出其羧基末端的甘氨酸残基。LC3-I再被Atg 7活化,转运至第2种E2样酶Atg 3,并被修饰成膜结合形式的LC3-II。LC3-II作为评价自噬强度的指标,在本研究的I/R组中表达较Sham 组明显上调,IPC 组比I/R 组表达更多。推测IPC可通过适量增强自噬强度,为心肌细胞提供更多能量,从而发挥心肌保护作用。其机制有待深入研究。
既往研究已证实自噬可通过抑制溶酶体Na+/H+质子泵,减少胞内Na+浓度,继而减少Na+/Ca2+交换,防止细胞内钙超载[9],避免线粒体通透性转换孔(Mitochordrial Permeability Transition Pore,mPTP)开放,从而减少促凋亡物质如细胞色素C(Cytochrome C,Cyt C)、凋亡诱导因子(Apoptosis Induced Factor,AIF)等的释放,减少心肌细胞凋亡。自噬对凋亡的抑制作用还与自噬体包裹损伤的线粒体有关,因而不但阻止了Cyt C 释放入细胞质,而且可以抑制凋亡小体的形成[10]。
自噬的调控通路可分为mTOR(Mammalian Target of Rapamycin)调节途径和mTOR 不依赖性调节途径[11]。在缺血缺氧等饥饿状态下,自噬的发生主要由mTOR 途径调控,自噬体的形成依赖于III型磷脂酰肌醇三磷酸激酶(Class III Phosphatidylinositol 3-Kinase,Class III PI3K)的作用[12]。Class III PI3K 抑制剂3-甲基腺嘌呤(3-MA)、渥曼青霉素可激活mTOR,由此干扰或阻断自噬小体的形成,减少细胞自噬发生。本实验在IPC 前用渥曼青霉素抑制自噬后,IPC的心肌保护作用被减弱,表明自噬可在IPC减少I/R 损伤中发挥重要作用。
1 Murry CE,Jennings RB,Reimer KA.Preconditioning with ischemia:a delay of lethal cell injury in ischemic myocardium[J].Circulation,1986,74(5):1124~1136.
2 刘剑刚,张蕾,史君鹤,等.预适应和后适应对缺血/再灌注损伤大鼠心肌组织炎症因子的影响[J].微循环学杂志,2011,21(3):4~7.
3 Gottlieb RA,Finley KD,Mentzer RM Jr.Cardioprotection requires taking out the trash[J].Basic Res Cardiol,2009,104(2):169~180.
4 Huang C,Liu W,Perry CN,et al.Autophagy and protein kinase C are required for cardioprotection by sulfaphenazole[J].Am J Physiol Heart Circ Physiol,2010,298(2):H570~579.
5 Hara T,Nakamura K,Matsui M,et al.Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice[J].Nature,2006,441(7095):885~889.
6 Downey JM,Krieg T,Cohen MV.Mapping preconditioning's signaling pathways:an engineering approach[J].Ann N Y Acad Sci,2008,1123:187~196.
7 Decker RS,Wildenthal K.Lysosomal alterations in hypoxic and reoxygenated hearts.I.Ultrastructural and cytochemical changes[J].Am J Pathol,1980,98(2):425~444.
8 Kim SJ,Peppas A,Hong SK,et al.Persistent stunning inducesmyocardial hibernation and protection:flow/function and metabolic mechanisms[J].Circ Res,2003,92(11):1233~1239.
9 Karwatowska-Prokopczuk E,Nordberg JA,Li HL,et al.Effect of vacuolar proton ATPase on pHi,Ca2+,and apoptosis in neonatal cardiomyocytes during metabolicinhibition/recovery[J].Circ Res,1998,82(11):1139~1144.
10 Meléndez A,Tallóczy Z,Seaman M,et al.Autophagy genes are essential for dauer development and life-span extension in C.elegans[J].Science,2003,301(5638):1387~1391.
11 Meijer AJ,Codogno P.Signalling and autophagy regulation in health,aging and disease[J].Mol Aspects Med,2006,27(5-6):411~425.
12 Petiot A,Ogier-Denis E,Blommaart EF,et al.Distinct classes of phosphatidylinositol 3'-kinases are involved in signaling pathways that controlmacroautophagy in HT-29cells[J].J Biol Chem,2000,275(2):992~998.
猜你喜欢
科学24小时(2021年4期)2021-03-22 02:31:09
传染病信息(2021年6期)2021-02-12 01:52:08
新课程·下旬(2017年11期)2018-01-22 21:37:11
中成药(2017年12期)2018-01-19 02:06:27
——可作为磷酸盐库再利用!
蔬菜(2018年12期)2018-01-16 05:27:32
福建轻纺(2017年12期)2017-04-10 12:56:39
现代检验医学杂志(2016年3期)2016-11-15 01:59:34
法医学杂志(2015年4期)2016-01-06 12:36:34
中国医疗美容(2015年4期)2015-04-27 02:24:07
现代检验医学杂志(2014年3期)2014-02-02 02:42:21
