
复方氟尿嘧啶注射液包封率的测定方法研究
2013-08-29 13:19张迪姜梅郭宏伟李沫李尚颖孙苓苓辽宁省食品药品检验所沈阳003沈阳市药品检验所沈阳000
中国药房 2013年29期
张迪,姜梅,郭宏伟,李沫,李尚颖,孙苓苓#(.辽宁省食品药品检验所,沈阳003;.沈阳市药品检验所,沈阳 000)
脂质体作为药物载体具有使药物带有靶向性、降低药物毒性、提高药物稳定性等特点[1],而其包封率是评价这类制剂质量的重要指标。常用的脂质体与游离药物的分离方法有:葡聚糖凝胶过滤法[2-3]、超速离心法[4]、超滤膜过滤法、超滤离心法等,而本文的研究对象来自某企业研制的复方氟尿嘧啶注射液为一种多相脂质体,准确测定其包封率有一定的困难。为建立其科学、合理的包封率测定方法,笔者试验了超滤离心法测定该制剂的包封率,同时进行了方法学研究。结果表明,建立的方法操作简单,结果准确、重复性好。
1 材料
1.1 仪器
2010型高效液相色谱(HPLC)仪(日本岛津公司);CP225D型十万分之一分析天平(德国Sartorius公司);高速低温离心机(日本日立公司)。
1.2 药品与试剂
复方氟尿嘧啶注射液(A企业,小试样品,批号:20100601、20100602、20100603,规格:10ml∶40mg);氟尿嘧啶对照品(中国食品药品检定研究院,批号:100187-200602,纯度:100.0%);甲醇为色谱纯。
2 方法与结果
2.1 色谱条件[5]
用十八烷基硅烷键合硅胶为填充剂(200mm×4.6mm,5μm);以水(0.05mol/L磷酸溶液调节pH 值至3.5)-甲醇(95∶5)为流动相,流速为1.0ml/min;紫外检测波长为265nm;柱温为35℃;进样量为10μl。
2.2 线性关系考察
精密称取氟尿嘧啶对照品9.73mg,置于25ml量瓶中,加甲醇15ml使溶解,加水稀释至刻度,摇匀;精密量取0.1、0.2、0.5、1、2、5ml,分别置于10ml量瓶中,加流动相稀释至刻度,摇匀。分别精密量取10μl注入色谱仪,记录峰面积。以氟尿嘧啶峰面积(y)为纵坐标、质量浓度(x)为横坐标进行线性回归,得y=3×106x+33186(r=0.9999,n=6)。结果表明,氟尿嘧啶检测质量浓度线性范围为3.892~194.6μg/ml。
2.3 精密度试验
取同一对照品溶液进样,每次10μl,连续进样6次,测定氟尿嘧啶峰面积RSD=0.1%。
2.4 专属性试验
按处方取相当于10ml注射液的其他原辅料(除氟尿嘧啶外),置于100ml量瓶中,加流动相稀释至刻度,摇匀,滤过。取续滤液,进样测定。结果表明,其他原辅料对氟尿嘧啶的测定无干扰,色谱见图1A。

图1 高效液相色谱图A.辅料;B.供试品;C.对照品;1.氟尿嘧啶Fig 1 HPLC chromatogramsA.recipients;B.test sample;C.substance control;1.fluorouracil
2.5 对空白脂质体透过能力的考察
取空白脂质体适量,分别置于100kD和3kD截留分子质量的超滤离心管中,10000r/min离心30min,采用HPLC-蒸发光散射(ELSD)法测定续滤液中磷脂酰胆碱的含量[6]。结果表明,滤液中均未检测到磷脂酰胆碱,说明2种超滤离心管均可以完全截留脂质体,且离心方法不会破坏脂质体。
2.6 游离药物的回收率
精密量取空白脂质体(A企业提供,取除氟尿嘧啶外的原辅料,照生产工艺同法制备)10ml,置于具塞试管中,加入相当于处方量50%、100%和150%的氟尿嘧啶对照品各3份,精密称定,反复翻转至氟尿嘧啶全部溶解。用100kD截留分子质量的超滤离心管,10000r/min离心30min。各精密量取续滤液1ml,置于100ml量瓶中,加流动相稀释至刻度,摇匀。精密量取滤液10μl注入色谱仪,记录色谱图,以外标法计算回收率。结果表明,超滤离心管对样品无吸附,回收率符合规定,详见表1。
2.7 截留分子质量的选择
取“2.6”项下试验样品,另采用3kD截留分子质量的超滤离心管,同法操作,计算回收率。结果表明,3kD截留分子质量的超滤离心管也对测定结果无影响,详见表1。说明2种超滤离心管均可。本方法选用100kD的超滤离心管。
2.8 重复性试验
精密量取本品1ml,共6份,用100kD截留分子质量的超滤离心管,10000r/min离心30min。精密量取滤液1ml,置于100ml量瓶中,加流动相稀释至刻度,摇匀。精密量取10μl注入色谱仪,记录色谱图,结果峰面积的RSD=0.8%。表明本方法重复性良好。
2.9 药物总量的测定[7]
精密量取本品1ml,置于100ml量瓶中,加甲醇10ml使溶解,用流动相稀释至刻度,摇匀。经0.45μm滤膜滤过后取续滤液10μl注入色谱仪,记录色谱图。
2.10 包封率测定方法
取本品,用100kD截留分子质量的超滤离心管,10000r/min离心30min;精密量取滤液1ml,置于100ml量瓶中,加流动相稀释至刻度,摇匀。取10μl注入色谱仪,记录色谱图,得峰面积为A;同时精密量取本品1ml,照“2.9”项下方法进样测定,得峰面积为B,按公式[(B-A)/B×100%]计算包封率。
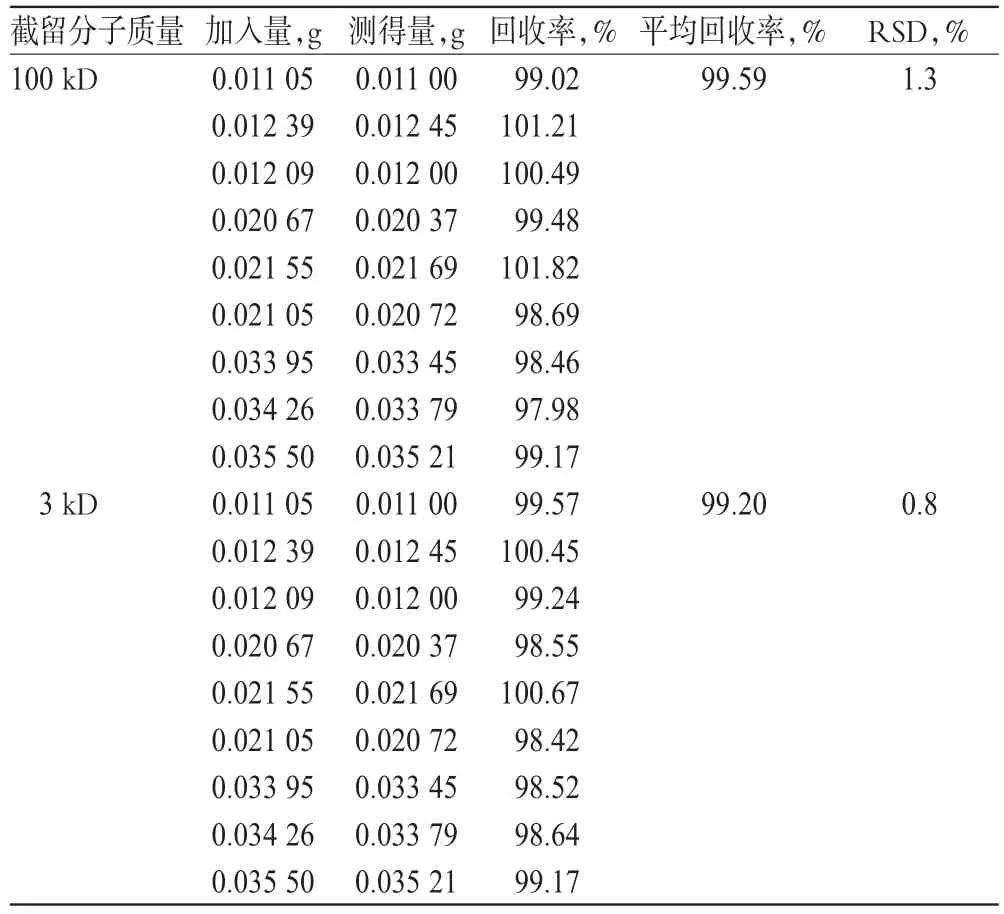
表1 2种超滤离心管条件下的回收试验结果(n=9)Tal 1 Results of recovery tests by 2kinds of ultrafiltration centrifuge tube(n=9)
2.11 2种方法包封率的测定
(1)按照“2.10”项下方法,取复方氟尿嘧啶注射液适量,测定包封率。结果,3批样品包封率分别为9.1%、8.8%和8.7%,对照品及供试品色谱图见图1B、C。
(2)采用原标准方法[8],精密量取本品10ml,采用超速低温离心法[18000r/min,温度(10±2)℃]离心15min,吸取脂质体层,置于10ml量瓶中,加水稀释至刻度,摇匀;精密量取1ml,置于50ml量瓶中,用甲醇-水(1∶5)溶液稀释至刻度,摇匀,照HPLC法[2010年版《中国药典》(二部)附录ⅤD][5]试验。用十八烷基硅烷键合硅胶为填充剂,以甲醇-水-三乙胺(16∶84∶0.2)为流动相,用80%磷酸溶液调节pH值至4.0,检测波长为266nm。取10μl注入色谱仪,记录色谱图,得峰面积为A′;同时精密量取本品1ml,置于50ml量瓶中,按上述方法自“用甲醇-水(1∶5)的溶液稀释至刻度”起,同法操作,得峰面积为B′,按公式(A′/B′×100%)计算[8]包封率分别为30.8%、31.6%和28.3%。
3 讨论
由于氟尿嘧啶为亲水性物质,在生产及贮存过程中易发生渗漏现象。该品种的含量测定结果[7]符合质量要求,但包封率均不高,而采用本方法测定其包封率也仅约为10%。
原标准[8]采用超速低温离心法,经试验,采用该方法无法将本品的外水相和脂质体完全分离,上清液不澄清;且离心后脂质体间易包裹部分外水相,导致测定的结果偏高(约30%),无法准确地反映产品质量。如果选用葡聚糖凝胶过滤法,脂质体在分子筛间筛选的过程中容易破裂,致使已包裹在脂质体中的药物溢出至外水相,导致包封率测定结果偏低,甚至是零。故最终选定超滤离心法作为测定方法。
超滤离心法是以压差为动力的膜分离技术,利用特定截留分子质量的滤膜使游离药物被过滤而脂质体被截留,从而使脂质体与游离药物分离。通过对空白脂质体透过能力的考察,结果表明该方法可完全截留脂质体,且不会破坏脂质体,保证了测定结果的可靠性。游离药物的回收率试验结果表明,该方法不会对游离药物造成吸附,可准确测定游离药物的含量。
综上所述,本文建立的方法可方便、准确地测定复方氟尿嘧啶注射液的包封率。
[1] 崔福德.药剂学[M].1版.北京:中国医药科技出版社,2002:495-496.
[2] 吴骏,朱家壁.阿昔洛韦脂质体包封率的测定[J].药物分析杂志,2003,23(3):213.
[3] 高晓黎,季兴梅.葡聚糖凝胶色谱柱法测定脂质体包封率的条件筛选[J].中国药学杂志,2003,38(7):515.
[4] 徐斌,赵晶.川穹嗪聚乳酸纳米粒的包封率测定[J].南京中医药大学学报,2011,27(1):77.
[5] 国家药典委员会.中华人民共和国药典:二部[S].2010年版.北京:中国医药科技出版社,2010:540-541、附录ⅤD.
[6] 张迪,杨错,孙宽,等.HPLC-ELSD法测定复方氟尿嘧啶注射液中磷脂酰胆碱的含量[J].中国药事,2012,26(9):974.
[7] 张迪,郭宏伟,孙宽,等.HPLC法测定复方氟尿嘧啶注射液含量及有关物质[J].中国药师,2012,15(3):356.
[8] 国家药典委员会.国家药品标准:化学药品地方标准上升国家标准:第七册[S].北京:人民卫生出版社,2002:138-139.
猜你喜欢
中国药学药品知识仓库(2022年13期)2022-07-03
药品评价(2021年17期)2021-11-06
临床与实验病理学杂志(2021年7期)2021-09-06
科技视界(2020年26期)2020-09-24
科技视界(2020年17期)2020-07-30
化工管理(2020年18期)2020-07-15
科技视界(2019年25期)2019-11-19
中国新技术新产品(2019年5期)2019-05-21
丝路视野(2018年8期)2018-05-14
化学教学(2018年1期)2018-02-28
