
铈钛混合氧化物负载磷钨酸吸附-分解NOx 性能
2013-08-20 00:51程琳王睿
无机化学学报 2013年6期
程 琳 王 睿
(山东大学环境科学与工程学院,济南 250100)
多酸催化剂具有稳定的阴离子结构和可调变的催化性能,兼具适宜的酸性和氧化还原性,是一种公认的环境友好型催化剂,一直以来都受到学术界和工业界的广泛关注[1-4]。Yang 等[5]发现,NO 通过取代磷钨酸(H3PW12O40,简记为HPW)二级结构中的结晶水而被吸附至HPW 二级结构中,在吸附温度为150 °C、 空速为5 000 h-1的条件下,HPW 可以吸附气流中70%的NO,吸附饱和后,经150 °C·min-1的程序升温,68.3%被吸附的NO 被还原成N2。Herring[6-7]、Kiennemann[8]及Moffat 等[9-10]也 对 杂 多 化合物用于NOx的吸附乃至随后的脱附或分解进行了报道,但迄今采用杂多化合物的文献中关于NOx催化分解过程中氧的去向,一直未见确凿的报道。Moffat 等[11]在其研究报道中不仅再次确认固态的HPW 能够有效去除NOx,并且证实HPW 可以把NOx吸附到其内部分子结构中,他们得到了NO 和NO2的协同吸附曲线,认为这项工作展现了该催化剂良好的应用前景。但在实际应用中,单纯杂多酸比表面积较小,机械强度低,热稳定性差,难以重复利用。将杂多酸负载于适合的载体上,可以提高比表面积和热稳定性,故而成为目前研究的热点。常用的氧化 物 载 体 包 括SiO2[12]、SnO2、TiO2[13-14]、TixZr1-xO4[15]及CexZr4-xO8[16],但迄今我们尚未见采用CeO2-TiO2作为HPW 的载体材料的文献报道。
CeO2具有优良的氧储存释放特性,但热力学稳定性较差,高温条件下易烧结,为提高其热力学稳定性及氧储存释放功能,可将其它过渡金属引入到CeO2晶体结构中,当CeO2和过渡金属氧化物结合后,其氧化还原性能和催化性能都会发生较大改变。本文报道了以CeO2-TiO2为载体负载HPW 对NOx的吸附-分解性能,并通过检测发现了氧的脱附;文中就HPW 负载量、负载方法、吸附温度等因素对催化剂吸附NOx效率的影响进行了系统研究;探讨了NOx分解过程中产物组成及N2收率与分解过程中升温速率之间的关系; 对吸附分解过程机理进行了探讨。
1 实验部分
1.1 主要原料和试剂
实验所用试剂均为分析纯。磷钨酸(H3PW12O40·nH2O)和CeCl3·7H2O 均购自天津科密欧化学试剂有限公司,所用H3PW12O40·nH2O 均置于烘箱内80 ℃条件下烘干12 h,以得到H3PW12O40·6H2O。TiCl4购于国药集团化学试剂有限公司,氨水(NH3·H2O)购于莱阳康德化学试剂有限公司。
1.2 铈钛氧化物的制备
制备过程参考文献[17]。称取物质的量的比为1∶1的CeCl3·7H2O 和TiCl4,分别加入250 mL 去离子水,配成溶液,将2 种溶液混合,边搅拌边逐滴滴加稀氨水,调节终点pH 值至8.0。将所得沉淀用去离子水多次洗涤,去除里面多余的阴离子。过滤,滤饼置于烘箱中105 ℃烘干12 h,所得产物置于管式炉中500 ℃煅烧5 h,所得催化剂标记为CeO2-TiO2。
1.3 HPW/CeO2-TiO2 催化剂系列的制备
1.3.1 PW/CTO
采用机械研磨法负载HPW。称取一定质量的CeO2-TiO2和HPW,于玛瑙研钵中研磨30 min,所得产物置于150 ℃下预处理2 h,由此法制得的催化剂HPW/CeO2-TiO2记为xPW/CTO(x 代表HPW 的质量分数,x=20%,40%,60%),催化剂研磨至-40+60目(0.250~0.420 mm)备用。
1.3.2 60%PW/CTOs
采用等体积浸渍法负载HPW。称取4 g CeO2-TiO2和6 g HPW 置于烧杯中,加入20 mL 去离子水,超声分散1 h,磁力搅拌过夜,于70 ℃下烘干,所得固体于150 ℃下预处理2 h,由此法制得的催化剂HPW/CeO2-TiO2记为60%PW/CTOs,催化剂研磨至-40+60 目(0.250~0.420 mm)备用。
1.4 HPW/CeO2-TiO2 吸附-分解NOx 与再生性能评价

图1 NOx 吸附性能评价实验装置图Fig.1 Schematic diagram of the setup for NOx adsorption
NOx吸附实验装置流程如图1 所示。催化剂吸附NOx性能实验在石英管固定床反应器上进行。所用气体为NO 稀释气,它是由99.99%的NO(体积分数)和N2配制而成。吸附实验以高纯N2为载气,采用质量流量计精确控制气路中气体流量,采用TH-990S 型NOx分析仪检测气路中的NO 和NO2浓度。通过NOx吸附效率和吸附容量评价催化剂NOx吸附性能,NOx吸附效率表达为(Co-Ce)/Co×100%,其中Co代表反应器进气气流中的NOx浓度,Ce代表经催化剂吸附后反应器出口气流中的NOx浓度;NOx吸附容量表达为NOads/m,其中NOads代表催化剂在最佳吸附温度下吸附NOx的质量,m 为实验中使用催化剂的质量,NOx吸附容量的单位为mg NOx/g(sorbent)。
实验过程中反应器进气气流中的NOx浓度为1 mL·L-1,O2体积分数8.8%,水蒸气体积分数为4.5%。催化剂吸附NOx后,将其用于TPD-MS 实验中(升温速率50 ℃·min-1,升温区间150~450 ℃),采用LZL-204 型质谱仪(北京分析仪器厂)检测其催化分解产物,并计算产物中N2的收率,其中,N2收率(YN2)是生成物N2的量(2AN2)与通过氮守恒计算的反应混合物含N 总量(ANO+2AN2+2AN2O)的比值。
1.5 催化剂的表征
采用傅立叶转换红外光谱仪(FTIR,Nicolet Avatar 370 型,美国)对样品进行红外表征,分辨率2 cm-1,测量波数范围400~4 000 cm-1,样品经KBr压片,样品与KBr 比例约为1∶50;采用X 射线衍射仪(XRD,西门子D-5000 型,德国)对样品进行XRD表征,以Cu 靶Kα(λ=0.154 06 nm)为辐射源,加速电压40 kV,电流40 mA,测试时采用步进扫描模式,步长0.08°,扫描速度0.2°·s-1,扫描范围10°~80°(2θ),测量X-射线强度的探测器为LynxEye 半导体阵列探测器; 采用全自动比表面积及孔隙度分析仪(BET surface area,康塔公司Quadrasorb SI 型,美国)测试催化剂的比表面积,通过BET 法计算样品总比表面积;采用冷场发射扫描电子显微镜(SEM,日立S-4800 型,日本)对样品形貌进行表征,工作电压5.0 kV,测试距离8 mm;采用原位漫反射红外-拉曼光谱仪(in situ DRIFTS,Thermo Nicolet NEXUS 670 傅立叶变换红外-拉曼光谱仪,美国)对HPW 催化分解NOx的过程进行了原位漫反射红外表征,测量波数范围400~4 000 cm-1,分辨率4 cm-1,附件为高温高压漫反射固体池,样品经KBr 压片,实验过程中升温区间为20~450 ℃,升温速率为2 ℃·s-1,温度升高至450 ℃后稳定3 min,然后降温至150 ℃,降温速率为5 ℃·s-1。
2 结果与讨论
2.1 傅立叶红外光谱表征
图2 为通过不同负载方法制备的HPW/CeO2-TiO2系列催化剂的红外谱图。等体积浸渍法和机械研磨负载法制得的催化剂在1 100~600 cm-1之间均出现归属于Keggin 结构的4 个特征峰,具体为:在1 080、983 及890 和804 cm-1处分别出现归属于PO、W=O 和W-O-W 的特征峰,这和Yang 等[5]的报导相吻合。此外,上述催化剂均在1 683 和3 300 cm-1处出现结晶水的弯曲振动峰。
2.2 X 射线衍射表征

图2 HPW/CeO2-TiO2 系列催化剂的红外谱图Fig.2 IR spectra for HPW/CeO2-TiO2 catalysts

图3 X 射线衍射图Fig.3 XRD patterns of the catalysts (*stands for CeO2;# stands for TiO2)
铈钛混合氧化物载体及其负载型催化剂HPW/CeO2-TiO2系列的XRD 图如图3 所示。由衍射线a可以看出,CeO2-TiO2的XRD 图中呈现典型的CeO2晶体的衍射峰,伴随着强度较弱的TiO2锐钛矿晶相,与文献[17]中报道的一致。衍射线b、c 和e 为CeO2-TiO2通过机械研磨法负载不同量的HPW 所得催化剂的XRD 图,其XRD 图均出现了载体和HPW的衍射峰,且对于不同负载量的催化剂,HPW 的衍射峰强度随着负载量的增加而增强,而载体的衍射峰强度则相应减弱,衍射峰的锐度变小。衍射线c 和d 为CeO2-TiO2分别通过机械研磨法和等体积浸渍法负载等量HPW 后的XRD 图,两者均出现了HPW 的衍射峰,但机械研磨法较等体积浸渍法所得催化剂的HPW 及载体中衍射峰强度都更强,这可能是由于通过等体积浸渍法制备催化剂时,在浸渍液的媒介作用下,HPW 与载体表面发生了化学作用,导致HPW 晶体结构被破坏,结晶度变差;也可能是由于浸渍法制备过程中,部分HPW 以弥散分布的微晶粒子簇形式高度均匀的分散至载体表面所致[18]。结合红外谱图(图2),40%PW/CTOs 仍保留了HPW 的Keggin 结构的特征峰,且P-O、W=O 和WO-W 未发生红移或蓝移,由此可知,HPW 与载体表面未发生化学作用,因此我们推测造成浸渍法所得催化剂中HPW 衍射峰强度较弱的原因可能是:浸渍过程中,部分HPW 以弥散分布的微晶粒子簇形式高度均匀的分散至载体表面所致。
我们通过立方晶系布拉格方程

计算H3PW12O40和CeO2-TiO2的晶胞尺寸,结果如表1 所示,其中hkl 代表晶面指数,d 代表晶面间距,a 代表晶胞参数。H3PW12O40的平均晶胞参数为1.22 nm,与文献[18-19]中描述相一致,CeO2-TiO2的平均晶胞参数为0.52 nm,由此可知CeO2-TiO2的晶胞参数小于H3PW12O40。

表1 H3PW12O40 和CeO2-TiO2 晶胞尺寸Table 1 Cubic lattice constants of H3PW12O40 and CeO2-TiO2
2.3 扫描电镜观测
我们对H3PW12O40和CeO2-TiO2进行了SEM 表征,结果如图4 所示。图4A 中CeO2-TiO2颗粒较大,呈不规则多面体形状,表面相对光滑; 图4B 中H3PW12O40形状不规则、 表面粗糙、 有层状突起,与CeO2-TiO2相比H3PW12O40颗粒尺寸更小。由此可见,在二次团聚形成的颗粒中,CeO2-TiO2颗粒尺寸较大,H3PW12O40颗粒尺寸较小。

图4 催化剂的扫描电镜图片Fig.4 SEM images of the catalysts
通过不同负载方法制备的催化剂的SEM 图片如图4C 和4D 所示。图4C 显示,在机械研磨过程中,CeO2-TiO2和H3PW12O40的颗粒尺寸均发生了不同程度的减小,这可能是机械研磨过程中机械力作用所致,H3PW12O40分散于CeO2-TiO2的周围,且分散较均匀; 催化剂40%PW/CTOs 的SEM 照片(图4D) 显 示,经 过 等 体 积 浸 渍 后,CeO2-TiO2和H3PW12O40的颗粒尺寸仍然较大,这可能是由于浸渍过程中颗粒团聚所致,等体积浸渍法所得的催化剂中H3PW12O40在CeO2-TiO2周围分散不均匀。
2.4 比表面积
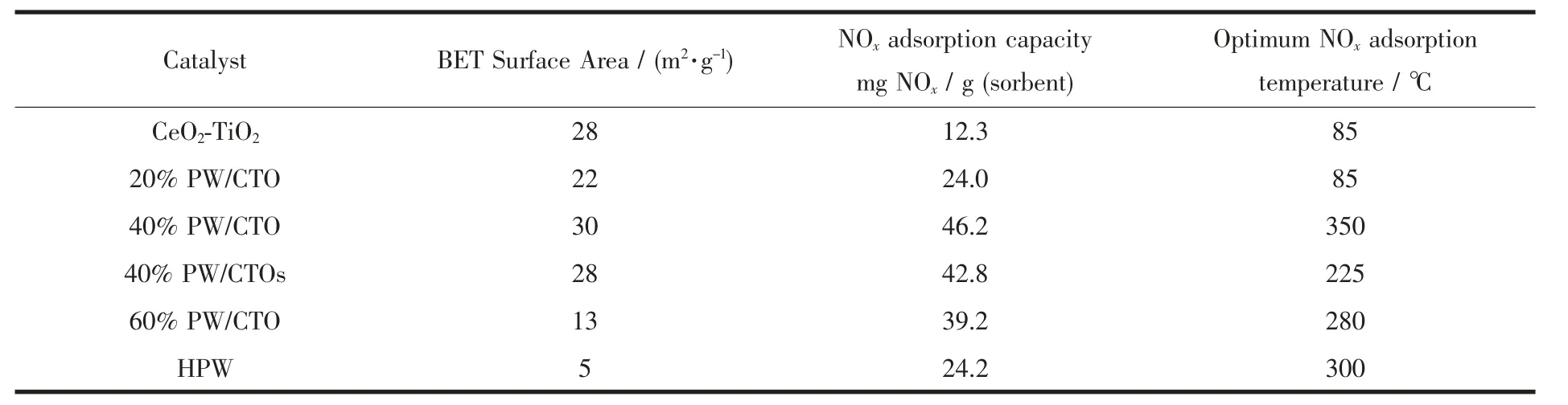
表2 比表面积和NOx吸附容量的关系Table 2 Relation between BET surface area and NOx adsorption capacity
表2 同时给出了CeO2-TiO2、HPW 及不同HPW负载量的HPW/CeO2-TiO2的比表面积测定结果。其中机械研磨法制备的催化剂的比表面积明显高于单一HPW,且随着负载量的增加,催化剂的比表面积出现先升后降的趋势,负载量为40%时达到最大值(30 m2·g-1),当负载量增至60%时,催化剂的比表面积下降至13 m2·g-1。机械研磨法和等体积浸渍法负载等量的HPW 后,前者的比表面积略大于后者。由表2 还可以看到,随负载量的变化,催化剂的NOx吸附容量呈现与其比表面积相同的递变规律,这表明两者之间存在正相关关联。
2.5 机械研磨法和等体积浸渍法所得催化剂吸附NOx 效率的比较
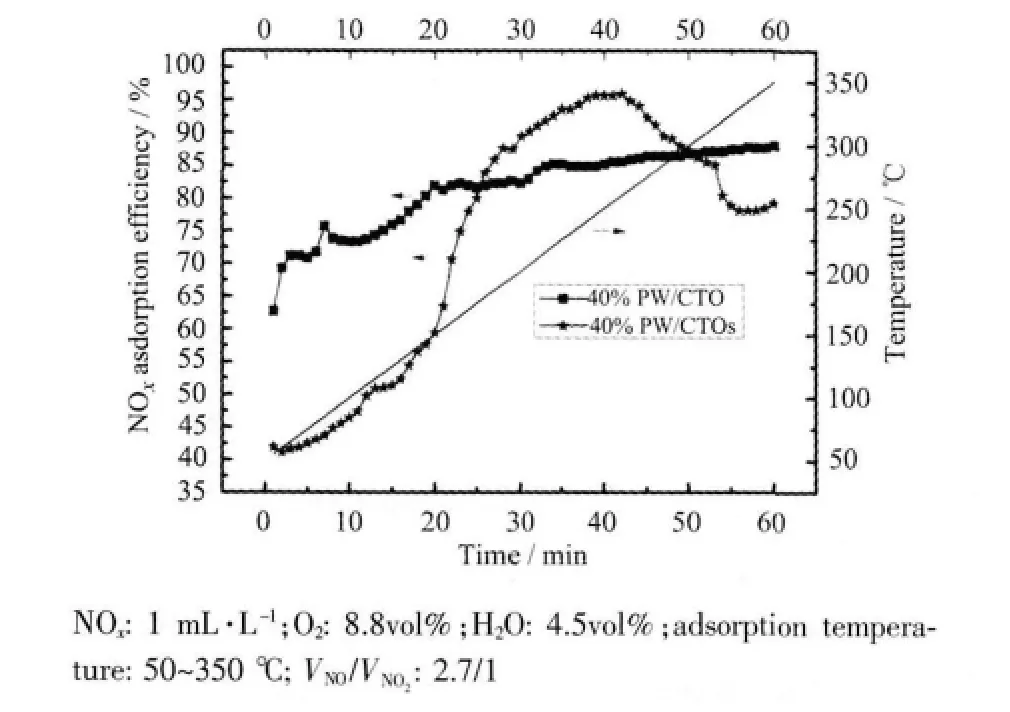
图5 不同负载方法吸附NOx 效率比较Fig.5 NOx adsorption efficiency obtained with catalysts prepared by different means
为比较2 种不同负载方法对所得催化剂吸附NOx性能的影响,我们将相同负载量下不同负载方法所得催化剂分别用于NOx吸附实验中,结果如图5 所示。可以看出,机械研磨负载法所得催化剂吸附NOx的性能略高于等体积浸渍法,尽管40% PW/CTOs 的最高吸附效率高于40% PW/CTO,但整体看来,后者的吸附性能更优越,且如表2 所示,后者的NOx吸附容量为46.2 mg NOx/g(sorbent),高于前者的42.8mgNOx/g(sorbent)。表2 显示40%PW/CTO 的比表面积略大于40%PW/CTOs(表2),这可能是造成2种负载方法所得催化剂脱硝效率出现差别的原因。
2.6 HPW 负载量对NOx 吸附效率的影响
考虑到机械研磨法制备的催化剂的性能优越性,我们进一步考察了HPW 负载量对机械研磨法所得催化剂吸附NOx效率的影响,测试了xPW/CTO(x=0%,20%,40%,60%)的NOx吸附性能,结果如图6 所示。

图6 HPW 负载量对NOx 吸附效率的影响Fig.6 Effect of HPW loading on NOx adsorption efficiency
图6 显示HPW 负载量是影响催化剂吸附NOx效率的一个重要因素。在0~40%的负载量范围内,催化剂对NOx的吸附效率随负载量增加而上升,并在负载量为40%时达到NOx吸附效率的最大值(90%);HPW 负载量较低时,HPW 活性吸附位数量较少,故而低负载量时,催化剂对NOx的吸附效率较低;随HPW 负载量的增加,吸附NOx的活性组分随之增加,对NOx的吸附效率也随之上升;表2 显示在0~40%的负载量范围内,催化剂的比表面积随负载量增加而逐渐增大,这也可能是造成在0~40%的负载量范围内,NOx吸附效率随HPW 负载量增加而上升的原因之一。当负载量增大到60%时,NOx的吸附效率出现了较大的下降,这可能是由于过高的负载量导致比表面积下降造成的,表2 显示当负载量升至60%时,比表面积降至13 m2·g-1。
机械研磨负载HPW 后所得到的催化剂均呈现出较高的NOx吸附效率,其最佳吸附效率均高于载体和HPW(60%)的最佳吸附效率,这表明负载HPW后,载体和HPW 对NOx的吸附存在协同作用,分析可能的原因如下:首先,CeO2-TiO2负载HPW 后所得负载型催化剂的比表面积较HPW 的比表面积均有较大提高,大的比表面积更有利于吸附剂对气体NOx的吸附; 其次,NO 只有在与NO2共存的条件下才会被HPW 吸附,且当VNO2∶VNO=1∶1 时,HPW 吸附NOx效率最高[11],CeO2-TiO2具有优良的储氧功能[17],可释放氧形成氧空位,这些活性氧将NO 迅速氧化为NO2,生成的NO2携同NO 被负载于其上的HPW 高效吸附; 再次,CeO2-TiO2在制备过程中,500 ℃的煅烧作用可能使得部分TiO2被引入CeO2晶格中,CeO2以萤石结构存在,Ti4+同晶取代Ce4+,晶格中的负电荷由邻近的正离子稳定,若是H+即产生B 酸中心[17],负载HPW 后,晶格中的负电荷由HPW 中的H+稳定,从而有效产生B 酸中心,增强了HPW 的B 酸强度,杂多酸对NOx的吸附效率与其B 酸酸强度成正比[10],因此负载HPW 后,NOx吸附效率得到提升。
2.7 吸附温度对催化剂吸附NOx 效率的影响
吸附温度是影响催化剂吸附NOx效率的一个重要因素。从图5 和图6 中均可以看出,除20%PW/CTO 外,实验范围内较高的温度更有利于催化剂对NOx的吸附,20%PW/CTO 的脱硝效率随温度的变化趋势与载体一致,这可能是因为HPW 负载量较低时脱硝效率主要受载体脱硝效率控制所致。
2.8 NOx 的催化分解
选取HPW 及40%PW/CTO 进行NOx催化分解实验。HPW 及40%PW/CTO 吸附NOx至近于饱和后,将其从150 ℃快速升温至450 ℃,升温速率分别为30、40 及50 ℃·min-1,并通过TPD-MS 检测产物组成及含量,结果见图7 和表3。
如图7 所示,吸附于HPW 和40%PW/CTO 上的NOx在快速升温过程中均发生了分解,产物为N2、O2和N2O(升温速率为50 ℃·min-1时,40%PW/CTO 的分解产物中没有N2O),另有部分NO 在高温下发生脱附,实验中未发现NO2的产生。
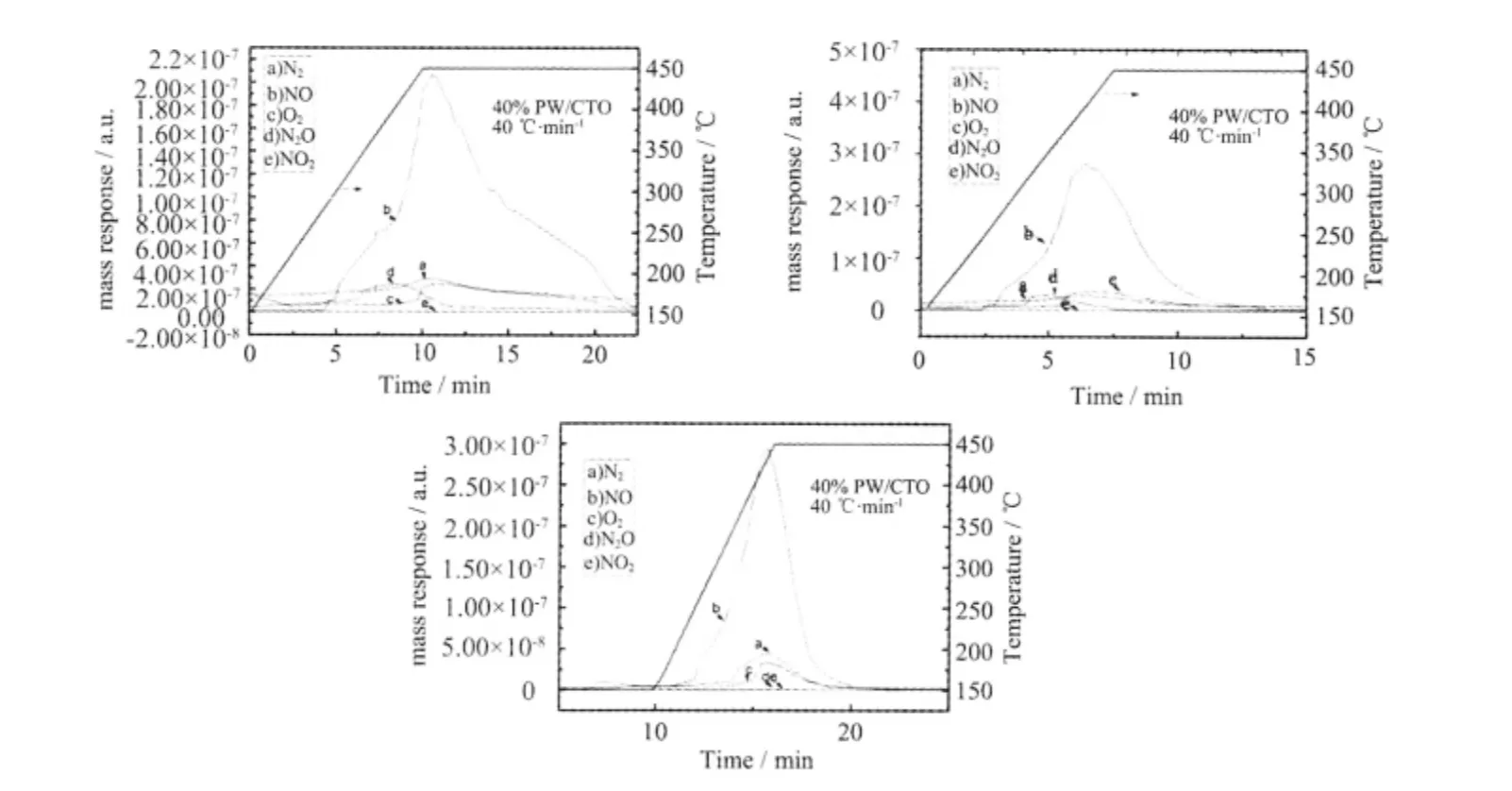
图7 40%PW/CTO 催化分解NOx的TPD-MS 检测结果Fig.7 TPD-MS profiles of NOx decomposition over 40%PW/CTO at different heating rates
表3 显示,不同的升温速率下,40%PW/CTO 的分解产物略有不同。随升温速率的加快,分解产物中N2O 的量逐渐减少,升温速率为50 ℃·min-1时,产物中不再有N2O 的生成;相应地,升温速率越快,N2收率越高,升温速率为30、40 和50 ℃·min-1时,对应的N2收率分别为11.4%、17.5%和30.5 %。分析原因可能为,瞬时高温下,活性N 优先相互结合形成N2,也即NOx分解为N2需要大的升温速率,因此通过更快的升温速率可望实现更高的N2收率。同时我们还比较了相同的升温速率(50 °C·min-1) 下HPW与40%PW/CTO 的N2收率,结果发现40%PW/CTO的N2收率高于HPW,前者为30.5%,后者为26.7%。

表3 HPW 和40%PW/CTO 对NOx的催化分解效果Table 3 Results of NOx decomposition over HPW and 40%PW/CTO
实验发现,O2的产生时间滞后于N2和N2O (图7),推测NOx在催化分解过程中,N-O 键发生断裂,生成活性N 和活性O,活性N 直接相互结合形成N2,另一部分活性N 与脱附的NO 结合形成N2O,而活性O 与HPW 的端氧W=O 及桥氧W-O-W 结合,并连接在配原子W 上,形成不稳定的过氧化多酸化合物中间体并滞留于催化剂上[19-20],在NOx高温分解的后期中间体上的O 脱落,并相互结合以O2形式逸出,因此,氧气的产生时间滞后于N2和N2O。
由于活性氧带有孤电子,如果它吸附在W=O键和W-O-W 键(包括W-Ob-W 和W-Oc-W,其中:Ob为角共用氧,Oc为边共用氧)上,活性氧会将电子传递给W=O 和W-O-W,使其电子云密度增大,键变短,能量变高,在红外谱图中将会表现为W=O 键和W-O-W 键的蓝移,在活性氧脱附时,活性氧结合成O2逸出,W-O 电子云密度恢 复,W=O 键和W-O-W键将红移至原出峰位置。为验证上文中对NOx分解过程中O2产生机理的推测,我们采用原位漫反射红外技术对HPW 催化分解NOx过程进行了表征,结果如图8 和表4 所示。
图8 中显示,吸附NOx后,HPW 在2 267 cm-1有一伸缩振动峰,归属于NOH+[6-7,18],在1 096、1 017、916、860 cm-1处分别出现归属于P-O、W=O、W-Ob-W、W-Oc-W 的特征峰[1]。
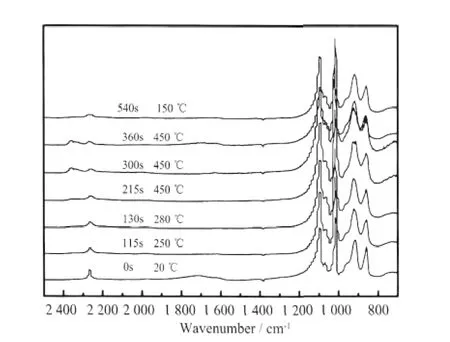
图8 HPW 催化分解NOx 过程的原位漫反射红外谱图Fig.8 In situ DRIFTS spectra of HPW catalyzed NOx decomposition process
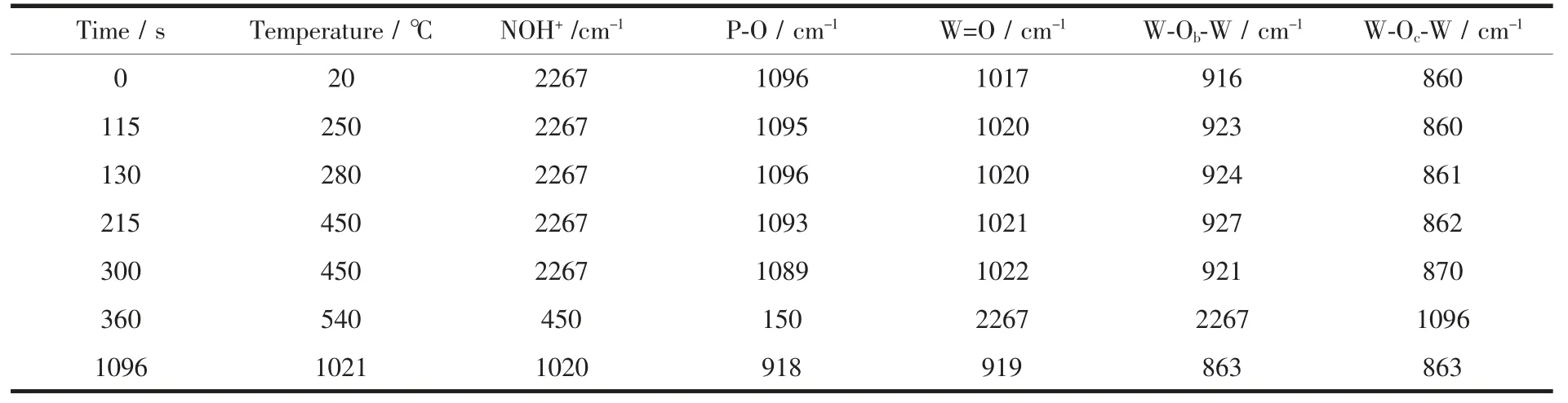
表4 HPW 催化分解NOx过程的原位漫反射红外结果Table 4 In situ DRIFTS results of HPW in NOx decomposition procedure
快速升温催化分解NOx过程中,2 267 cm-1处归属于NOH+的振动峰随温度升高呈现峰强度变弱、峰面积减小的趋势,说明快速升温过程中吸附于催化剂上的NOx逐渐消失,结合TPD-MS 对分解产物的检测结果,推断NOx发生分解,NOH+中N-O 键断裂,产生N2,O2和N2O。表4 显示,NOx催化分解反应前期(快速升温至450 ℃的过程中),HPW 红外谱图中W=O、W-Ob-W、W-Oc-W 均发生蓝移,P-O 键发生红移,具体为:W=O 由1 017 cm-1蓝移至1 022 cm-1,W-Ob-W 由916 cm-1蓝移至927 cm-1,W-Oc-W由860 cm-1蓝移至870 cm-1,说明W 的电子云密度变大,W=O 和W-O-W 的键能变强;HPW 中P-O 键发生红移,由1 096 cm-1红移至1 089 cm-1,图8 显示,该过程磷钨酸Keggin 结构中外围W3O13中的W=O 和W-O-W 均发生蓝移,表明W 的电子云密度增大,W=O 和W-O-W 键长均变短,这可能引起中心P-O 键长变长,P 原子周围电子云密度减小,从而导致P-O 键能减弱,最终表现为P-O 键的红移。
催化分解NOx后期,催化剂中W=O、W-Ob-W、W-Oc-W 均发生了红移,红移后的谱图和催化分解NOx前的漫反射红外谱图相似,表明O2脱附后,WO 电子云密度逐渐恢复;P-O 键蓝移,蓝移后的谱图和催化分解NOx前的漫反射红外谱图相似。降温至150 ℃后,在仪器误差范围内,催化剂的漫反射红外谱图和催化反应前一致,进一步表明,NOx催化分解反应完成后,催化剂的结构得到了恢复。
综上,HPW 催化分解NOx过程的原位漫反射红外结果验证了我们对NOx催化分解过程中O2产生机理推测的合理性。
2.9 催化剂的再生与重复利用
Hodjati 等[21]指 出,H2O 与NOx之 间 存 在 竞 争 吸附,反应温度低于100 ℃时,HPW 主要吸附水蒸气,当反应温度高于100 ℃时,主要吸附NOx,他们还据此提出了催化剂冷却脱附再生新方法: 催化剂吸附NOx后,在湿空气(水蒸气体积分数为5%)中逐渐冷却,可将NOx完全脱附,这种脱附方法避免了程序升温脱附过程中可能造成的HPW 受热分解问题,提高了HPW 的重复利用效率。基于此原理,我们采取低温通入水蒸气的方法对使用后的催化剂进行再生,通入的水蒸气体积分数为10%,再生温度为25 ℃,再生时间为2 h,再生后的催化剂重新用于NOx吸附实验,结果如图9 所示。催化剂再生后,其重复利用效果较理想,实验发现,再生后的催化剂对NOx的吸附容量达到45.5 mg NOx/g (sorbent),其催化分解NOx的N2收率达29.6%(升 温速率50 ℃·min-1)。由此可见,对上述催化剂而言,通水蒸气是一种较为有效的再生方式。
2.10 催化剂吸附-分解NOx 及再生过程的红外分析
我们对吸附NOx前后、催化分解NOx及再生后的催化剂40%PW/CTO 进行了傅立叶红外表征,结果如图10 所示。谱线a 显示,吸附NOx前40%PW/CTO 在1 100~600 cm-1之间出现了归属于Keggin 结构的特征峰:P-O(1 080 cm-1)、W=O(983 cm-1)、W-O-W(888 和799 cm-1)。吸附NOx后,谱线b 中归属 于Keggin 结构的特征峰不变,说明吸附NOx前后催化剂Keggin 结构未发生变化;2 270 cm-1出现归属于NOH+中N-O 的振动峰,和文献[6-7,18]报道的一致;红外谱图中未发现铈钛混合氧化物吸附NOx的证据。
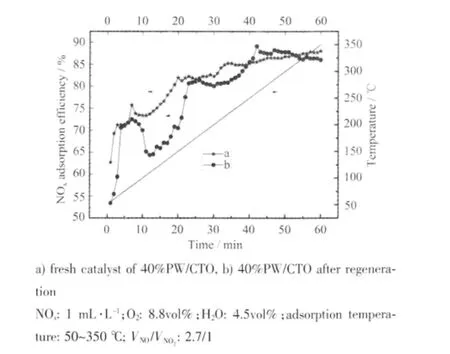
图9 再生前后催化剂吸附NOx 效率Fig.9 Efficiency of NOx adsorption for catalysts before and after regeneration
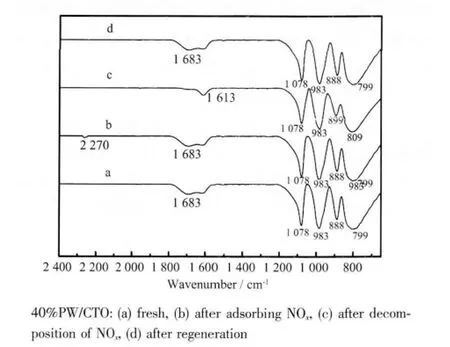
图10 铈钛氧化物负载磷钨酸吸附分解NOx 前后红外谱图Fig.10 FTIR spectra of catalysts before and after NOx decomposition, as well as after regeneration
谱线c 为催化分解NOx后40%PW/CTO 的红外谱图,与谱线b 相比,2 270 cm-1处归属于N-O 的振动峰消失,说明高温作用下,吸附于催化剂上的NOx发生分解;NOx分解后,催化剂在1 100~600 cm-1之间归属于HPW 的Keggin 结构的4 个特征峰得到了保留;桥氧键W-O-W 的出峰位置由888 和799 cm-1分别蓝移至895 cm-1和809 cm-1,与原位漫反射红外的结果一致(图8);催化剂在1 683 cm-1处的H2O的弯曲振动峰红移至1 613 cm-1,表明NOx分解后HPW 内结晶水数目发生改变,在高温分解NOx的过程中催化剂二级结构发生变化; 通入含有水蒸气的空气再生后的催化剂的红外谱图(谱线d)则与吸附分解NOx之前的催化剂的红外谱图(谱线a)完全一致,在1 683 cm-1处出现H2O 的弯曲振动峰,证明通入含有水蒸气的空气可有效补充HPW 在NOx分解过程中丢失的结晶水。
3 结 论
(1) 相比于等体积浸渍法,机械研磨法更适合CeO2-TiO2负载H3PW12O40,后者所制备的系列催化剂对NOx具有较高的吸附效率,均高于H3PW12O40及载体本身的脱硝效率。
(2) 在0~60%的负载量范围内,随着H3PW12O40负载量的增加,机械研磨法制备的催化剂吸附NOx的效率呈现先升后降趋势,最佳负载量为40%,对应的最高NOx吸附率为90%。
(3) 当温度从150 ℃升高至450 ℃时,被催化剂吸附的NOx发生分解,分解产物组成和N2收率均受升温速率的显著影响。升温速率为30 ℃·min-1和40 ℃·min-1时,分解产物为N2、O2和N2O;当升温速率为50 ℃·min-1时,产物中只有N2和O2,不再有N2O; 升温速率越快,N2的收率越高; 升温速率为50 ℃·min-1时,NOx分解产物中N2的收率达30.5%,明显高于单一H3PW12O40。
(4) 在NOx吸附过程中,NOx和H3PW12O40上的质子生成NOH+;在NOx的分解过程中,N-O 键发生断裂,生成活性N 和活性O,一部分活性N 相互结合形成N2,另一部分活性N 与脱附的NO 结合形成N2O,而活性O 与HPW 的端氧W=O 及桥氧W-O-W结合并连接在配原子W 上,形成不稳定的过氧化多酸化合物中间体并滞留于催化剂上,在NOx高温分解的后期中间体上的O 脱落,并相互结合以O2形式逸出。向吸附-分解NOx后的催化剂床层通入含有水蒸气的空气,可有效补充NOx分解过程中H3PW12O40失去的结晶水,从而恢复催化剂优良的NOx吸附-分解性能,实现催化剂的有效再生利用。
[1] WANG En-Bo(王恩波), HU Chang-Wen(胡长文), XU Lin(许 林). Introduction to Polyoxometalates Chemistry (多 酸 化学导论). Beijing: Chemical Industry Press, 1998.
[2] WANG Xiao-Lan(王晓兰), LI Yang-Guang(李阳光), MENG Jing-Xin(孟 靖 昕), et al. Chinese J. Inorg. Chem. (Wuji Huaxue Xuebao), 2010,26(3):391-397
[3] ZHAO Zhi-Feng(赵志凤), ZHOU Bai-Bin(周百斌), SU Zhan-Hua(苏 占 华), et al. Chinese J. Inorg. Chem. (Wuji Huaxue Xuebao), 2009,25(5):900-905
[4] WANG Jing-Ping(王敬平), WU Qiang(吴 强), NIU Jing-Yang(牛 景 杨). Chinese J. Inorg. Chem. (Wuji Huaxue Xuebao), 2002,18(9):957-960
[5] Yang R T, Chen N. Ind. Eng. Chem. Res., 1994,33:825-831
[6] Herring A M, McCormick R L. J. Phys. Chem. B, 1998,102(17):3175-3184
[7] Herring A M, McCormick R L, Boonrueng S R. J. Phys.Chem. B, 2000,104(19):4653-4660
[8] Hodjati S, Vaezzadeh K, Kiennemann A, et al. Top. Catal.,2001,16/17(1/2/3/4):151-155
[9] Bonardet J L, Fraissard J, Mc Garvey G B, et al. J. Catal.,1995,151:147-154
[10]Belanger R, Moffat J B. J. Mol. Catal. A: Chem., 1996,114:319-329
[11]Belanger R, Moffat J B. Environ. Sci. Techno., 1995,29:1681-1685
[12]McCormick R L, Boonrueng S K, Herring A M. Catal. Today,1998,42:145-157
[13]Thomas S, Vaezzadeh K, Pitchon V. Top. Catal., 2004,30/31:207-213
[14]Kozhevnikov I V. Catalysts for Fine Chemical Synthesis Vol.2: Catalysis by Polyxometalates. West Sussex: John Wiley &Sons Ltd, 2002.
[15]Gómez-García M A,Pitchon V,Kiennemann A.Catal.Today,2005,107/108:60-67
[16]Gómez-García M A, Pitchon V, Kiennemann A. Environ.Sci. Technol., 2005,39:638-644
[17]Benjaram M R, Ataullah K, Pandian L. J. Phys. Chem.B, 2005,109:3355-3363
[18]ZHANG Xue-Yang(张学杨), CHENG Lin(程琳), YANG Feng(杨 烽), et al. Chem. J. Chin. Univ.(Gaodeng Xuexiao Huaxue Xuebao), 2012,33(8):1826-1834
[19]Chen N, Yang R T. J. Catal., 1995,157:76-86
[20]Hiskia A, Papaconstantinou E. Inorg. Chem., 1992,31:163-167
[21]Hodjati S, Petit C. J. Catal., 2001,197:324-334
猜你喜欢
化学工程师(2023年1期)2023-02-17
世界农药(2022年10期)2022-11-10
当代化工研究(2022年11期)2022-06-27
选煤技术(2022年2期)2022-06-06
石材(2022年1期)2022-05-23
能源化工(2021年2期)2021-12-30
理化检验-化学分册(2020年12期)2021-01-26
军事文摘(2020年18期)2020-10-27
石材(2020年2期)2020-03-16
上海农业科技(2019年1期)2019-02-22
