
DNA酶传感器的研究进展
2013-08-14 09:08王敏娟
化学与生物工程 2013年6期
王敏娟
(宝鸡文理学院化学化工学院,陕西 宝鸡721013)
DNA属于生物聚合物当中的一类,是最重要的遗传信息载体。20世纪90年代初,人们发现一部分DNA分子也表现出催化活性,称之为脱氧核酶(DNA酶,DNAzyme)。目前,DNA酶和适体统一被称为功能化的DNA,可以通过体外筛选、扩增技术得到。
虽然DNA酶有催化功能,但是当它单独存在时,其催化作用很弱;只有在可与其特异性结合的辅助因子共存时,才能激活DNA酶的活性而表现出催化性能,这些辅助因子包括氨基酸、核酸、金属离子和小的有机分子等。基于此原理,DNA酶被作为识别物质广泛应用于一系列金属离子(如 Hg2+、Cu2+、Pb2+等)的识别和检测。
1 DNA酶的特点
(3)与蛋白质相比,DNA酶发生多次的复性、变性之后,其结合力或者活性仍然不受影响,因此,可以在相当苛刻的条件下使用和储存。
(4)DNA 酶可以用于光纤和微阵列技术[3,4],可以用于实时或者多种金属离子的同时检测。
2 DNA酶的作用机理
与金属离子特异性识别的DNA酶由两部分组成:第一部分是特异性识别金属离子的环状部分;第二部分是与底物链杂交的部分。当底物链与酶链杂交之后,如果有与酶链特异性识别的金属离子存在,酶链就会被激活,底物链的rA处被水解而断裂。因此,DNA酶作为生物识别物质广泛应用于多种金属离子的检测。
DNA酶作为生物探针具有以下特点:
(1)DNA酶的制备不依赖于动物和细胞,而是可以通过体外筛选或指数富集配体系统进化技术来得到,而且其序列一旦确定,就可以通过化学的方法进行合成。因此,同批生产的DNA酶几乎没有批间误差,而且组成确定,纯度高。
(2)DNA酶是单链的DNA序列,比较容易合成,大约在1~2d内即可完成,而且比RNA的合成便宜。在生理条件下,DNA的稳定性比蛋白质高1000倍,比RNA高10万倍[1]。近期发现,DNA酶经常以球状蛋白的形式存在[2],所以不易被核酸内切酶识别。
3 DNA酶传感器在光学分析中的研究进展
金属中毒,是指人体内某种金属的含量过多而引起的慢性或急性中毒,并非过量摄入有害的金属离子才会导致人体中毒,人体需要的金属离子摄入过量也会导致金属中毒。通常情况下,重金属一旦摄入人体将无法排出体外。因此如何快速、准确地检测某种金属离子是否超标已经成为研究热点。
目前,金属离子的检测方法主要有原子吸收光谱法[5]、电 感 耦 合 等 离 子 体 质 谱 法[6-8]、阳 极 溶 出 伏 安法[9,10]、毛细管电泳法[11]等,这些方法 大多需要精密的仪器和熟练的操作人员,而且样品需要预处理,难以实现对金属离子的实时检测。因此人们建立了基于DNA酶的生物传感方法用于检测金属离子。下面就主要介绍DNA酶在光学传感器中的应用进展。
3.1 基于DNA酶的荧光传感器
荧光传感器的构建是基于分子识别物质与目标物作用前后体系荧光信号的变化来对目标物进行检测的。早在2000年,Li等[12]就筛选出了与Pb2+特异性结合的DNA酶(17E),同时构建了检测Pb2+的荧光传感器。在底物链(17DS)的5′端标记了荧光物质TAMRA,在酶链(17E)的3′端标记了猝灭剂 Dabcyl,当标记有荧光物质的底物链单独存在于溶液中时,体系的荧光信号很强;当加入酶链之后,由于底物链和酶链杂交成双螺旋结构,使得荧光物质和猝灭物质间发生了荧光共振能量转移(FRET),TAMRA的荧光被猝灭,体系荧光信号大大减弱;向体系中加入Pb2+后,Pb2+作为酶的辅助因子,使得酶的活性被激活,底物链在切割点被切断,荧光猝灭现象被消除,TAMRA的荧光信号恢复,体系荧光信号增强,此方法在4℃下的检出限为10nmol·L-1。虽然此方法能够高灵敏度、高选择性地检测Pb2+,但是必须在4℃下进行,如果温度升高到室温就会导致底物链从酶链上解旋下来而产生过高的背景信号。为了解决这个问题,Liu等[13]建立了一种基于分子内和分子间同时猝灭的双猝灭新型荧光检测方法来降低背景信号,原理如图1所示。
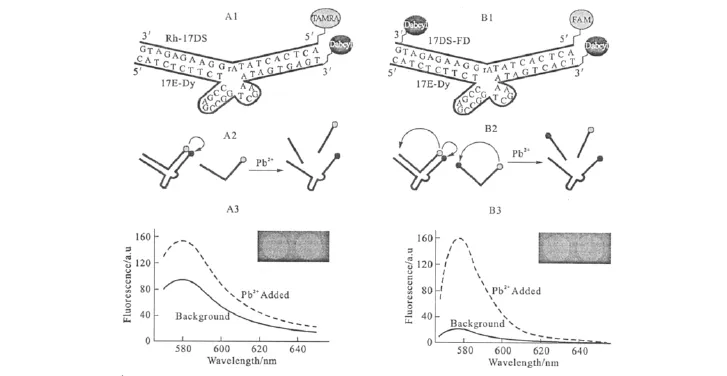
图1 基于DNA酶检测Pb2+的单猝灭(A)与双猝灭(B)荧光传感器对比Fig.1 Comparison of the single-(A)and double-quencher(B)DNAzyme-based Pb2+ sensor designs
由图1可看出,双猝灭效应对高的背景信号产生了明显的抑制作用,并且可以在较宽的温度范围内对Pb2+进行检测且不影响选择性。2005年,Swearingen等[14]又建立了非均相检测Pb2+的荧光法。将一端修饰有猝灭剂、另一端修饰有巯基的酶链(HS-17E-Dy)通过巯基自组装固定到金表面,再与标记有荧光物质的底物链(17DS-Fl)杂交,不存在Pb2+时,荧光基团与猝灭基团较接近,荧光信号被猝灭;与Pb2+反应后,底物链被切断,荧光信号恢复,同样根据加入Pb2+前后体系荧光信号的变化来对Pb2+进行定量检测。与均相荧光法相比,非均相的检测方法使得固定在金表面的酶链可以再生和长期保存,而且检出限(1nmol·L-1)比均相检出限降低了一个数量级。
虽然之前构建的基于DNA酶的金属离子传感器响应速度快,但是须对酶链或者底物链进行标记,因而对酶的活性有一定的影响,且成本较高。
2009年,Xiang等[15]将dSpacer位点引入到传感器的构建当中,他们在DNA双链区的酶链上引入了一个dSpacer位点,以荧光小分子2-氨基-5,6,7-三甲基-1,8-萘啶(ATMND)为信号物质,建立了基于DNA酶的无标记型荧光法用于检测Pb2+。单独的ATMND溶液的荧光信号很强,但是当它嵌入到有dSpacer位点的双链中形成配合物时,荧光信号被猝灭。Pb2+的存在使得酶链被激活而将底物链切断,双螺旋结构被破坏,小分子被释放出来,荧光信号增强,在优化的条件下,这种无标记的方法对溶液中4nmol·L-1的Pb2+也能产生特异性的响应。
2010年,Zhang等[16]又将高猝灭效率的分子信标模型和酶催化检测Pb2+模型联合起来,建立了一种检测Pb2+的催化分子信标(CAMB)新模型。此方法与前面的方法相比,有以下几个优点:第一,催化分子信标背景信号低,改变了信噪比,灵敏度比直线型的底物探针模型高;第二,分子信标的猝灭效率高,使得在操作中可以用比酶链浓度较大的底物链来达到信号放大的目的;第三,酶链无需修饰猝灭物质来起猝灭的作用,可以用来传感一系列的目标物(金属离子或有机分子)。该法检测限(600pmol·L-1)较基于DNA酶的Pb2+检测方法更低。
随后,Carmi等[17,18]和 Liu等[19]筛选出了与 Cu2+特意性识别的DNA酶。Liu等[20]采用降低背景信号的双猝灭方法,构建了检测Cu2+的荧光传感器,该传感器可以对溶液中不低于35nmol·L-1(2.3ppb)的Cu2+进行检测,且响应速度特别快,在2~4min内就可以完成检测。此外,Liu等[21,22]还运用DNA酶构建了一系列用于检测其它金属离子(如Uo2+、Hg2+)的荧光传感器。
3.2 基于DNA酶的光度传感器
光度传感器的构建是基于分子识别物质与目标物作用前后体系吸光度的变化进行定量检测,或者根据加入目标物前后,体系最大吸收波长的变化进而引起溶液颜色的变化,来进行目视检测,即所谓的比色法。
Liu等[22]在这方面也进行了一系列的工作,将之前构建的用于检测Pb2+的荧光传感器模型改为光度传感器[23]。该光度传感器是将底物链两端延伸,使得延伸的部分正好与自组装在纳米金粒子(13nm)上的单链DNA杂交,然后与酶链杂交,在没有与Pb2+作用之前,延伸的底物链将溶液中修饰有单链DNA的纳米金粒子连在一起,纳米金团聚,胶体溶液由酒红色变为蓝色;与Pb2+作用后,底物链被水解断裂,纳米金再度分散到溶液中,颜色由蓝色又变回酒红色,从而可根据颜色和吸光度的变化来检测Pb2+。
2008年,Lee等[23]采用检测Pb2+的比色法模型,构建了基于DNA酶的标记型和非标记型光度传感器用于检测UO,原理如图2所示。

图2 基于DNA酶的光度传感器用于检测UO的研究Fig.2 Scheme of labeled and lable-free colorimetric sensors based on Au nanoparticle in the absence and presence of UO
3.3 基于DNA酶的电化学发光传感器
近年来,电化学发光分析方法因高的灵敏度和可以重复多次检测等优点也受到了研究者的关注。Ma等[24]以钌联吡啶为电化学发光物质,构建了信号增强型电化学发光DNA酶传感器用于检测Pb2+的方法。将3′端标有钌联吡啶的与底物链杂交好的DNA酶固定在石墨电极的表面,在没有加入目标物Pb2+之前,酶链与底物链以双螺旋结构存在,由于双螺旋结构有一定的刚性,使得钌联吡啶与电极表面的距离增大;加入Pb2+后,Pb2+将底物链切断,DNA的双螺旋结构被打开,从而使得钌联吡啶与电极的距离缩小,电化学发光信号增强,根据体系电化学发光信号的变化对目标物Pb2+进行定量检测,检测限为1.4pmol·L-1。此方法也可以用于其它金属离子的检测。
4 结语
近年来,光学分析法由于灵敏度高、操作简单和检测费用低等特点而得到了迅速的发展。由于DNA酶合成较便宜、性质稳定、易于储存,而且在金属离子的存在下可以被激活而发生催化作用,因此,研究者们构建了一系列基于DNA酶的光学生物传感器用于金属离子的检测。DNA酶有望用于实时或者多种金属离子的同时检测。
[1]Breaker R R.Catalytic DNA:In training and seeking employment[J].Nat Biotechnol,1999,17(5):422-423.
[2]Nowakowski J,Shim P J,Prasad G S,et al.Crystal structure of an 82-nucleotide RNA-DNA complex formed by the 10-23DNA enzyme[J].Nat Struct Biol,1999,6(2):151-156.
[3]Walt D R.Techview:Molecular biology.Bead-based fiber-optic arrays[J].Science,2000,287(5452):451-452.
[4]Taylor L C,Walt D R.Application of high-density optical microwell arrays in a live-cell biosensing system[J].Anal Biochem,2000,278(2):132-142.
[5]Tahan J E,Granadillo V A,Romero R A.Electrothermal atomic absorption spectrometric determination of Al,Cu,Fe,Pb,V and Zn in clinical samples and in certified environmental reference materials[J].Anal Chim Acta,1994,295(1-2):187-197.
[6]Aggarwal S K,Kinter M,Herold D A.Determination of lead in urine and whole blood by stable isotope dilution gas chromatography-mass spectrometry[J].Clin Chem,1994,40(8):1494-1502.
[7]Liu H W,Jiang S J,Liu S H.Determination of cadmium,mercury and lead in seawater by electrothermal vaporization isotope dilution inductively coupled plasma mass spectrometry[J].Spectrochim Acta Part B:Atomic Spectroscopy,1999,54(9):1367-1375.
[8]Bowins R J,McNutt R H.Electrothermal isotope dilution inductively coupled plasma mass spectrometry method for the determination of sub-ng·mL-1levels of lead in human plasma[J].J Anal At Spectrom,1994,9(11):1233-1236.
[9]Feldman B J,Osterloh J D,Hata B H,et al.Determination of lead in blood by square wave anodic stripping voltammetry at a carbon disk ultramicroelectrode[J].Anal Chem,1994,66(13):1983-1987.
[10]Jagner D,Renman L,Wang Y.Determination of lead in microliter amounts of whole blood by stripping potentiometry[J].Electroanalysis,1994,6(4):285-291.
[11]Regan F B,Meaney M P,Lunte S M.Determination of metal ions by capillary electrophoresis using on-column complexation with 4-(2-pyridylazo)resorcinol following trace enrichment by peak stacking[J].J Chromatogr B:Biomed Appl,1994,657(2):409-417.
[12]Li J W,Lu Y.A highly sensitive and selective catalytic DNA biosensor for lead ions[J].J Am Chem Soc,2000,122(42):10466-10467.
[13]Liu J,Lu Y.Improving fluorescent DNAzyme biosensors by combining inter-and intramolecular quenchers[J].Anal Chem,2003,75(23):6666-6672.
[14]Swearingen C B,Wernette D P,Crogek D M,et al.Immobilization of a catalytic DNA molecular beacon on Au for Pb(Ⅱ)detection[J].Anal Chem,2005,77(2):442-448.
[15]Xiang Y,Tong A J,Lu Y.A basic site-containing DNAzyme and aptamer for label-free fluorescent detection of Pb2+and adenosine with high sensitivity,selectivity,and tunable dynamic range[J].J Am Chem Soc,2009,131(42):15352-15357.
[16]Zhang X B,Wang Z D,Xing H,et al.Catalytic and molecular beacons for amplified detection of metal ions and organic molecules with high sensitivity[J].Analytical Chemistry,2010,82(12):5005-5011.
[17]Carmi N,Breaker R R.Characterization of a DNA-cleaving deoxyribozyme[J].Bioorg Med Chem,2001,9(10):2589-2600.
[18]Carmi N,Balkhi H R,Breaker R R.Cleaving DNA with DNA[J].Proc Natl Acad Sci USA,1998,95(5):2233-2237.
[19]Liu J W,Lu Y.A DNAzyme catalytic beacon sensor for paramagnetic Cu2+ions in aqueous solution with high sensitivity and selectivity[J].J Am Chem Soc,2007,129(32):9838-9839.
[20]Liu J W,Brown A K,Meng X L,et al.A catalytic beacon sensor for uranium with parts-pertrillion sensitivity and millionfold selectivity[J].PNAS,2007,104(7):2056-2061.
[21]Liu J W,Lu Y.Rational design of"turn-on"allosteric DNAzyme catalytic beacons for aqueous mercury ions with ultrahigh sensitivity and selectivity[J].Angew Chem Int Ed,2007,46(40):7587-7590.
[22]Liu J W,Lu Y.A colorimetric lead biosensor using DNAzyme-directed assembly of gold nanoparticles[J].J Am Chem Soc,2003,125(22):6642-6643.
[23]Lee J H,Wang Z D,Liu J W,et al.Highly sensitive and selective colorimetric sensors for uranyl(UO):Development and comparison of labeled and label-free DNAzyme-gold nanoparticle systems[J].J Am Chem Soc,2008,130(43):14217-14226.
[24]Ma F,Sun B,Qi H,et al.A signal-on electrogenerated chemiluminescent biosensor for lead ion based on DNAzyme[J].Anal Chim Acta,2011,683(2):234-241.
猜你喜欢
云南化工(2021年6期)2021-12-21
昆明医科大学学报(2021年8期)2021-08-13
科学(2020年2期)2020-08-24
汽车电器(2019年1期)2019-03-21
中央民族大学学报(自然科学版)(2018年3期)2018-11-09
光谱学与光谱分析(2016年5期)2016-07-12
河南科技(2015年8期)2015-03-11
天津医科大学学报(2011年4期)2011-07-13
中国洗涤用品工业(2011年6期)2011-03-20
中国洗涤用品工业(2011年5期)2011-03-20
