
高效液相色谱法测定阿托伐他汀钙中的有关物质
2013-07-31 07:18王文清汪秋兰谢斌张光勋方建国施春阳
中国医院药学杂志 2013年19期
王文清,汪秋兰,谢斌,张光勋,方建国,施春阳
(1.华中科技大学同济医学院附属同济医院药学部,湖北武汉430030;2.湖北丝宝药业有限公司,湖北 武汉430019)
阿托伐他汀钙(atorvastatincalcium)属HMG-CoA还原酶抑制剂,本身无活性,口服吸收后的水解产物在体内竞争性地抑制胆固醇合成过程中的限速酶羟甲戊二酰辅酶A还原酶,使胆固醇的合成减少,也使低密度脂蛋白受体合成增加,中度降低血清三酰甘油水平和增高血高密度脂蛋白水平[1-3],临床上主要用于高胆固醇血症、动脉粥样硬化和冠心病的治疗[3-4]。本品为美国华纳兰伯特公司(后被辉瑞公司兼并)的专利产品,由于其疗效显著,不良反应小,耐受性好,随后陆续在全球65个国家和地区上市,销量连续数年雄居全球畅销药物的榜首。近年来,本品也实现国产,先后有7家制药企业获得批准文号[5]。欧洲药典7.1版(EP7.1)[6]和美国药典34版(USP34)[7]均收载本品,其有关物质的检查均选用醋酸铵-乙腈-四氢呋喃为流动相进行梯度洗脱;EP 7.1中的有关物质计算采用以主成分对照品为对照的外标法,USP34中有关物质计算为:已知杂质是以杂质对照品为对照的外标法,未知杂质按照峰面积归一化法。本实验选用醋酸铵-乙腈进行梯度洗脱,有关物质计算中,已知杂质采用杂质对照品法,未知杂质采用不加校正因子的主成分自身对照法,方法专属性强,重复性好,可更科学、准确地检测已知杂质和未知杂质的含量。
1 仪器与试剂
1.1 仪器 LC-20AD型高效液相色谱仪(日本岛津公司),包括LC-20AD泵,SIL-20A型自动进样器,SPD-M20A型二极管阵列检测器(190~800nm),LCsolution工作站;AUW220D型双量程分析天平(日本岛津公司)。
1.2 试剂 阿托伐他汀钙对照品(中国食品药品检定研究院,批号100590-200802,按无水阿托伐他汀钙计,供HPLC测定,本品含量为95.0%,供UV测定,本品含量为95.5%);阿托伐他汀钙原料药(浙江新东港药业股份有限公司,批号20100701,20100801,20101003);杂质对照品:杂质 A(批号G0J255)、杂质B(批号 G0J256)、杂质 C(批号G0J265)、杂质D(批号 G0J257)、杂质 H(批号 F0K120)、杂质 I(批号F0K121),均购自美国药典委员会;甲醇、乙腈、N,N-二甲基甲酰胺(美国天地公司,色谱纯);其他试剂均为分析纯,水为新制多效纯化水。
2 方法与结果
2.1 色谱条件 ApolloC18柱(250mm ×4.6mm,5 μm),流动相A为醋酸铵缓冲液:0.02mol·L-1的醋酸铵溶液,并用冰醋酸调节至 pH(4.5 ±0.05) - 乙腈(70∶30),流动相 B:乙腈,梯度洗脱(0~15min:A86%;15~30min:A86%→90%;30~50min:A90%→55%;50~68min:A55%;68~80min:A55%→25%;80~95min:A25%);检测波长:244nm;流速:1.1mL·min-1,柱温:40 ℃,进样量:20 μL。理论板数按阿托伐他汀峰计算不低于3000,阿托伐他汀主峰与各杂质峰分离度良好。
2.2 检测波长的选择 精密称取阿托伐他汀钙对照品及杂质对照品 A、B、C、D、H 和 I适量,用 N,N-二甲基甲酰胺溶解并稀释制成每1mL中约含阿托伐他汀和杂质各10μg的混合溶液,精密量取上述溶液20μL进样,分别在200~400nm波长范围内进行紫外扫描,结果各杂质对照品最大吸收值均在244nm附近,且阿托伐他汀钙在244nm波长处具有最大吸收峰,故选择244nm作为紫外检测波长。
2.3 溶液的配制
2.3.1 杂质对照品混合溶液 避光操作。精密称取阿托伐他汀杂质对照品 A、B、C、D、H和 I适量,用 N,N-二甲基甲酰胺溶解并稀释制成每1mL中约含各杂质对照品1μg的混合溶液,作为杂质对照品混合溶液。
2.3.2 系统适用性溶液 避光操作。称取阿托伐他汀钙对照品和6个杂质对照品(阿托伐他汀杂质 A、B、C、D、H、I)适量,用N,N-二甲基甲酰胺稀释制成每1mL中约含阿托伐他汀1mg、各杂质1μg的混合溶液,作为系统适用性溶液。
2.3.3 供试品溶液 避光操作。本品约25mg,精密称定置25mL棕色量瓶中,加N,N-二甲基甲酰胺适量振摇使溶解并稀释至刻度,摇匀即得。
2.3.4 对照溶液 精密量取上述供试品溶液适量,用N,N-二甲基甲酰胺稀释制成每1mL中约含阿托伐他汀1μg的溶液,作为0.1%对照溶液。
2.4 专属性试验
2.4.1 空白干扰试验 按上述色谱条件,精密量取系统适用性溶液、供试品溶液、空白溶剂各20μL进样,记录色谱图。由图1可见,各杂质峰与主峰之间分离度良好。
2.4.2 破坏试验 取本品约10mg5份,分别置5个10mL量瓶中,分别加2mLN,N-二甲基甲酰胺振摇使溶解,1号供试品量瓶中加2mol·L-1的盐酸溶液 0.5mL,摇匀,室温中放置12h后,以2mol·L-1的氢氧化钠溶液0.5mL 中和;2 号供试品量瓶中加入4mol·L-1的氢氧化钠溶液2mL,摇匀,室温放置12h,以4mol·L-1的盐酸溶液2mL 中和;3 号供试品量瓶中加入30%双氧水溶液0.5mL,摇匀,室温放置12h;4号供试品量瓶于4500Lx强光下破坏30h;5号供试品量瓶于80℃恒温烘箱中放置12h,向上述量瓶中加N,N-二甲基甲酰胺至刻度,摇匀,滤过,即得酸、碱、氧化、光照、高温破坏的供试品溶液。同法对空白溶剂分别进行上述破坏试验。结果显示:阿托伐他汀钙原料药经酸、碱、氧化、高温和光照破坏后,含量分别降至74.02%,99.16%,97.20%,88.23%,98.70%。表明本品经酸、高温破坏有明显降解,降解的主要产物为杂质H,且经破坏后,各杂质峰与阿托伐他汀主峰分离度良好,对测定无干扰。结果见图2。
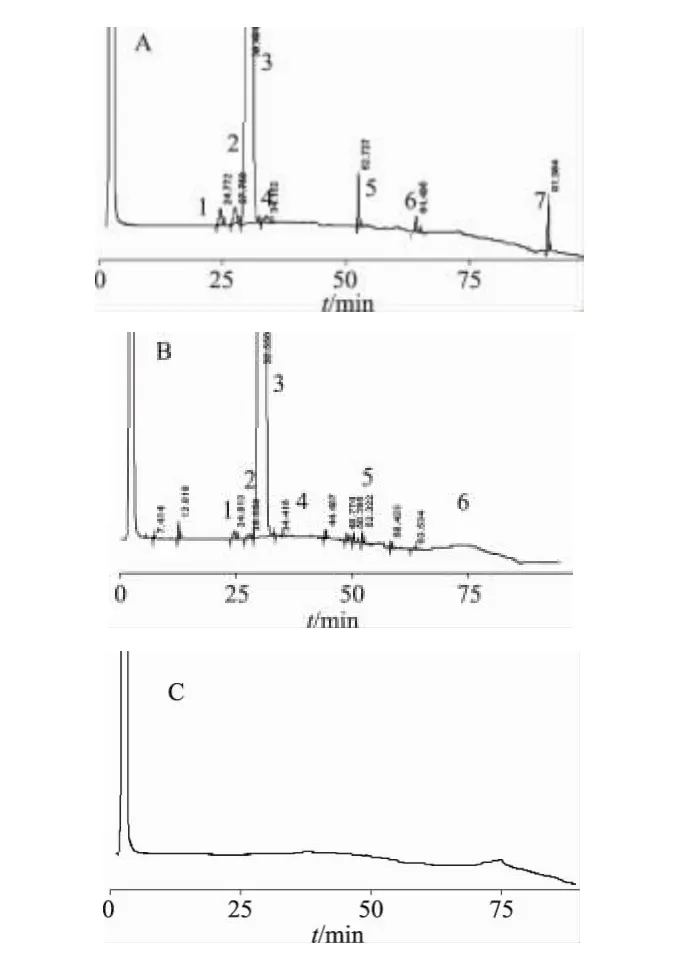
图1 高效液相色谱图A.系统适用性;B.供试品;C.空白溶剂1-杂质A;2-杂质B;3-阿托伐他汀钙;4-杂质C;5-杂质H;6-杂质D;7-杂质IFig1 HPLCchromatogramsA.Systemsuitability;B.Sample;C.blanksolvent1-impurityA;2-impurityB;3-atorvastatin;4-impurityC;5-impurity H;6-impurityD;7-impurityI
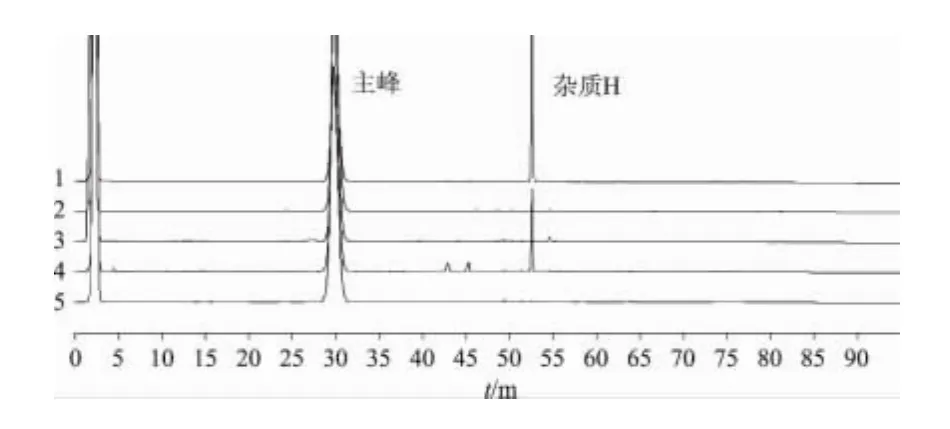
图2 阿托伐他汀钙破坏试验高效液相色谱图1-酸破坏;2-碱破坏;3-氧化破坏;4-高温破坏;5-光照破坏Fig1 AtorvastatincalciumHPLCchromatographsofdestructiontest1-treatedbyacid;2-treatedbybase;3-treatedbyoxidation;4-treatedby hightemperature;5-treatedbylight
2.5 相对响应因子的计算 精密称取杂质对照品A4.76 mg,B5.08mg,C4.78mg,D5.20mg,H4.97mg,I5.13mg,分别置10mL棕色量瓶中,用N,N-二甲基甲酰胺溶解并稀释至刻度,摇匀。作为杂质对照品混合溶液贮备液。再用N,N-二甲基甲酰胺稀释制成每1mL各约含10μg的杂质对照品混合溶液。同法制备每1mL约含10μg的阿托伐他汀钙对照品溶液,分别精密量取各杂质对照品混合溶液及阿托伐他汀钙对照品溶液各20μL,重复进样5次,记录色谱图。按峰面积/浓度计算各杂质与阿托伐他汀的相对响应因子。结果杂质 A、B、C、D、H 和 I的相对响应因子分别为0.94、1.02、0.94、1.03、1.07和0.94,各杂质的相对响应因子均在0.9~1.1之间。故在杂质对照品难以获得或价格较贵时,可采用不加校正因子的主成分自身对照法计算有关物质的含量。
2.6 定量限与检测限检查 分别精密量取杂质对照品混合溶液和阿托伐他汀钙对照品溶液各20μL进样,再不断用N,N-二甲基甲酰胺稀释后测定其峰面积,记录色谱图。以信噪比S/N=10时的进样量确定最低定量限,即阿托伐他汀钙最低定量限为0.528ng,杂质 A、B、C、D、H 和 I最低定量限分别为2.0900,3.5320,3.8260,4.1600,0.574,4.1080ng;以信噪比S/N=3时的进样量确定最低检测限,即阿托伐他汀钙最低检测限为0.264ng,杂质 A、B、C、D、H 和 I最低检测限 分 别 为 1.0450,1.7660,1.9120,2.0800,0.2870,2.0540ng。
2.7 线性关系 精密量取“2.5”项下杂质对照品混合溶液贮备液1mL,置25mL棕色量瓶中,用N,N-二甲基甲酰胺稀释至刻度。再精密量取0.1,0.25,0.5,1,1.5,2.5,5mL,分别置10mL棕色量瓶中,用N,N-二甲基甲酰胺稀释至刻度,摇匀,作为供试品溶液。分别取各供试品溶液20μL,进样,记录色谱图,以峰面积对浓度绘制标准曲线(表1)。
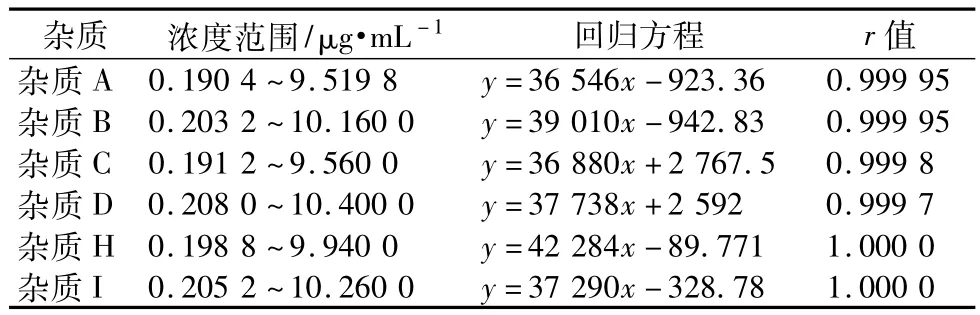
表1 线性范围、回归方程及相关系数结果Tab1 Theresultsoflinearrange,regressionequationandcoefficient
2.8 已知杂质定量限的精密度试验 精密量取已知杂质定量限项下的溶液20μL,连续进样5次,测得杂质A、B、C、D、H 和 I峰面积的 RSD 值分别为3.26%,3.25%,4.04%,3.07%,2.69%,1.21%。均小于 5%,即精密度较好。
2.9 重复性试验 取同一批(批号20100701)的供试品6份,按照“2.3”项下方法制备并按上述色谱条件进行测定,结果杂质 A、B、C、D、H 的含量平均值分别为0.10%(RSD=2.11%)、0.08%(RSD=3.26%)、0.03%(RSD=4.91%)、0.04%(RSD=3.47%)和0.05%(RSD=4.76%),杂质 I未检出,最大未知杂质为0.10%(RSD=1.79%),总杂质为0.54%(RSD=2.65%)。
2.10 溶液的稳定性考察
2.10.1 室温条件下杂质对照品混合溶液的稳定性 取杂质对照品混合溶液,于室温(20~25℃)条件下放置,分别于0,2,4,8,12,16,24h 定点精密取20 μL 进样,测得杂质 A、B、C、D、H 和 I峰面积的 RSD 值分别为0.47%,1.01%,2.72%,3.84%,0.36%,0.19%。说明杂质对照品混合溶液于在此条件下放置24h内较稳定。
2.10.2 室温条件下供试品溶液的稳定性 按照“2.3.3”项下方法处理供试品1份,于室温(20~25℃)下放置,分别于0,2,4,6,8,10h 定点精密吸取 20 μL 进样,测得杂质 A、B、C、D 和 H 峰面积的 RSD 值分别为1.70%、5.65%、5.11%、9.39%和12.17%,杂质 I未检出。说明供试品溶液于室温(20~25℃)条件下放置10h,杂质A较稳定,杂质B、C、D和H不稳定。
2.10.3 冷藏条件下供试品溶液的稳定性 供试品溶液于冷藏(2 ~8℃)条件下放置,分别于0、2、4、8、12、18、24h 定点精密吸取20μL进样,测得杂质A、B、C、D、H峰面积的RSD值分别为1.42%,2.78%,4.82%,3.91%,4.86%,杂质 I未检出。说明供试品溶液于冷藏(2℃ ~8℃)条件下放置24h,各已知杂质较稳定。
2.11 回收率试验 取已知杂质含量的本品适量(批号20100701,约相当于阿托伐他汀100mg),置100mL棕色量瓶中,用N,N-二甲基甲酰胺溶解并稀释至刻度,摇匀,滤过,作为供试品溶液,精密量取供试品溶液5mL于10mL量瓶中,加N,N-二甲基甲酰胺稀释至刻度,摇匀,作为空白溶液。另分别取上述供试品溶液5mL,加入9个10mL量瓶中,再分别精密量取杂质对照品混合溶液(杂质A、B、C、D、H、和I)(质量浓度分别为9.5198,10.1600,9.5600,10.4000,9.9400,10.2600 μg·mL-1)0.25,0.5,1mL 各 3 份,分别加入上述9个量瓶中,再用N,N-二甲基甲酰胺稀释至刻度,摇匀,作为回收率供试品溶液。精密量取上述溶液各20μL进样,记录色谱图,测定含量并计算回收率。结果杂质A、B、C、D、H、和 I的平均回收率分别为98.14%(RSD=2.05%),100.87%(RSD=4.47%),97.16%(RSD=5.19%),102.55%(RSD=3.45%),101.15%(RSD=5.16%),100.35%(RSD=3.12%)。
2.12 有关物质的检查方法 按“2.3”项下方法制备供试品溶液、0.1%对照溶液及已知杂质对照品混合溶液。精密量取对照溶液20μL,注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的20%。精密量取供试品溶液、对照溶液和杂质对照品混合溶液各20μL,分别注入液相色谱仪,记录色谱图。供试品溶液色谱图中如显杂质峰,杂质 A、B、C、H、D、I按外标法计算含量,均不得过0.3%,任一未知杂质峰面积不得大于对照溶液主峰面积的2倍(0.2%),总杂质不得过1.0%。
2.13 有关物质检查结果 对3批阿托伐他汀钙分别进行有关物质检查,结果见表2。
3 讨论
本试验测定的已知杂质包括杂质 A、B、C、D、H和 I,但其他已知杂质,如杂质E为对映异构体,需用正相HPLC法进行分离,杂质F和G,美国药典委员会目前暂未提供。其中杂质A、C、F和G为工艺杂质,杂质D、H和I为降解产物,杂质B和E为工艺及降解产物,了解杂质的来源可更好地控制本品的内在质量。
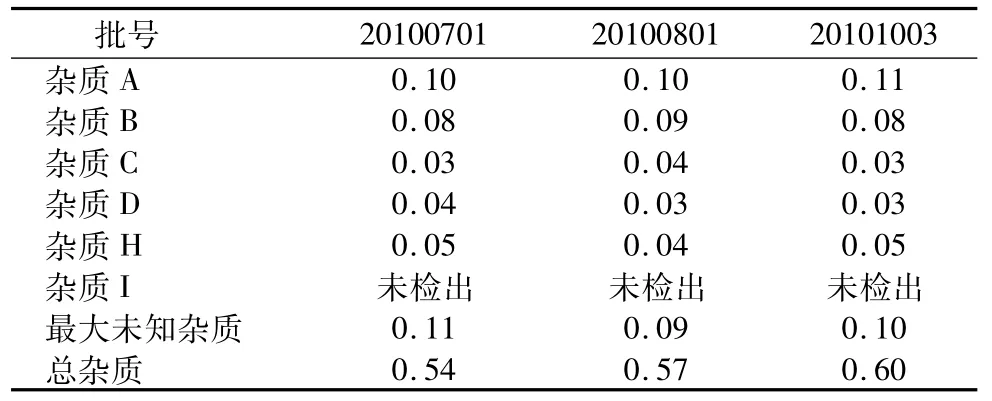
表2 样品有关物质测定结果(n=3,%)Tab2 Resultsofrelatedsubstancesofsample(n=3,%)
阿托伐他汀为多取代的吡咯衍生物,在甲醇中易溶,在水中极微溶解,结构中含有2个羟基的羧酸侧链,故显弱酸性,在流动相中加入弱酸性的缓冲液,以抑制阿托伐他汀的解离,增强其保留值。参考标准及文献[5-8]色谱条件,均选用醋酸盐缓冲液或枸橼酸盐缓冲液-乙腈-四氢呋喃进行梯度洗脱作为流动相,流动相中加入四氢呋喃,可有效地改善色谱峰峰型,但基线噪音很大且基线漂移,研究发现醋酸盐缓冲液的浓度对主成分峰及各杂质峰之间分离度无明显影响,故本试验将流动相进行适当调整,选用醋酸盐缓冲液(0.02 mol·L-1的醋酸铵溶液),并用冰醋酸调节至 pH(4.5 ±0.05)-乙腈进行梯度洗脱作为流动相。
从专属性试验考察结果可知,阿托伐他汀主峰tR约为30 min,但经详细的耐用性实验研究,发现流动相的比例及其pH值对本品的分离度及保留时间影响较大,故可通过增加或减少乙腈的比例或醋酸铵缓冲液的pH值来调整流动相,使阿托伐他汀主峰的保留时间控制在26~34min之间,以达到满意的分离度。
从破坏试验和供试品溶液稳定性考察的结果可知,本品在强酸、强氧化、光照、高温及溶剂中均不稳定,主要降解产物为杂质H(阿托伐他汀内酯),故本品供试品溶液需在避光条件下操作,于冷藏(2~8℃)条件下放置24h内测定或于室温(20~25℃)放置3h内测定[7]。
[1]SeanCSweetman主编,李大魁等译.马丁代尔大药典[M].35版.北京:化学工业出版社,2009:955.
[2]张象麟.药物临床信息参考[M].成都:四川科学技术出版社,2005:547-548.
[3]陈新谦.新编药物学[M].17版.北京:人民卫生出版社,2011:420.
[4]罗庆红,吴华春.阿托伐他汀对高胆固醇血症的疗效观察[J].中国医院药学杂志,2008,28(4):293-295.
[5]国家食品药品监督管理局.国家药品标准新药转正标准[S].[第36册]西药部分.标准编号WS1-(X-106)-2003Z:102.
[6]EP7.1 [S].2011:3380.
[7]USP34-NF29 [S].2011:1949.
[8]张亚平,杨淑莲,陈芬儿.HPLC法检测阿托伐他汀钙有关物质的含量及光学纯度[J].药物分析杂志,2010,30(12):2311-2313.
猜你喜欢
纺织标准与质量(2022年5期)2022-10-27
山东化工(2018年15期)2018-09-20
环境保护与循环经济(2017年6期)2018-01-22
大陆桥视野·下(2017年2期)2017-03-30
中国继续医学教育(2015年6期)2016-01-07
首都食品与医药(2015年18期)2015-11-03
中国当代医药(2015年16期)2015-03-01
河南医学研究(2014年5期)2014-02-27
河南医学研究(2014年3期)2014-02-27
药学研究(2012年2期)2012-10-25

- 中国医院药学杂志的其它文章
- 高效分子排阻色谱法测定注射用头孢西丁钠聚合物