
高效分子排阻色谱法测定注射用头孢西丁钠聚合物
2013-07-31 07:18苑华张冬宋家雄石家庄市食品药品检验所河北石家庄0500河北省食品药品检验院河北石家庄0500河北医科大学药学院河北石家庄05007
中国医院药学杂志 2013年19期
苑华,张冬,宋家雄 (.石家庄市食品药品检验所,河北石家庄0500;.河北省食品药品检验院,河北 石家庄0500;.河北医科大学药学院,河北石家庄05007)
头孢西丁(cefoxitin)是美国Merk公司研制的头孢菌素类半合成抗生素,抗菌活性和抗菌谱与第二代头孢菌素相同。它对革兰阴性菌如大肠杆菌、肺炎杆菌、吲哚阳性的变形杆菌和沙雷菌、克雷白杆菌、流感杆菌等均有较强的抗菌效果,对葡萄球菌和多种链球菌也有较好作用[1]。研究表明,β-内酰胺类抗生素引发的速发型过敏反应并非抗生素药物本身所致,而是与药物中存在的高分子聚合物有关[2]。注射用头孢西丁钠收载于中国药典2010年版二部,其标准项下未进行高分子聚合物的检查[3],鉴于此,我们采用TSKGELG2000SWXL色谱柱建立了高效分子排阻色谱法[4-5]测定注射用头孢西丁钠中聚合物的方法,与传统的凝胶色谱法比较,本法提高了分离效能,缩短了检验周期,检验结果也更为准确可靠。
1 材料
岛津LC-20AT高效液相色谱仪。头孢西丁对照品(中国食品药品检定研究院,130572-200701,含量:95.3%);注射用头孢西丁钠样品(样品1:A厂,批号11052004;样品2:B厂,批号11042421;样品3:C厂,批号110203);乙腈为色谱纯,其他试剂为分析纯。
2 方法与结果
2.1 色谱条件 色谱柱:TSK-GELG2000SWXL(5μm,7.8mm ×300mm);流动相:0.01mol·L-1磷酸盐缓冲液[0.01mol·L-1NaH2PO4溶液-0.01mol·L-1Na2HPO4(34∶66)]-乙腈(95∶5);检测波长:232nm;柱温:室温;进样量:20 μL;流速:0.8mL·min-1。
2.2 溶液的制备及测定方法 取本品适量,精密称定,加水溶解并稀释制成每1mL中含头孢西丁0.5mg的溶液,作为供试品溶液(临用新制);精密量取供试品溶液1mL,置100 mL量瓶中,加水稀释至刻度,作为对照溶液。精密量取供试品溶液与对照品溶液各20μL,注入液相色谱仪,记录色谱图;供试品溶液色谱图中如有杂质峰,保留时间小于头孢西丁峰的各杂质峰面积的和不得大于对照溶液主峰面积的1.5倍(1.5%)(图 1,2)。
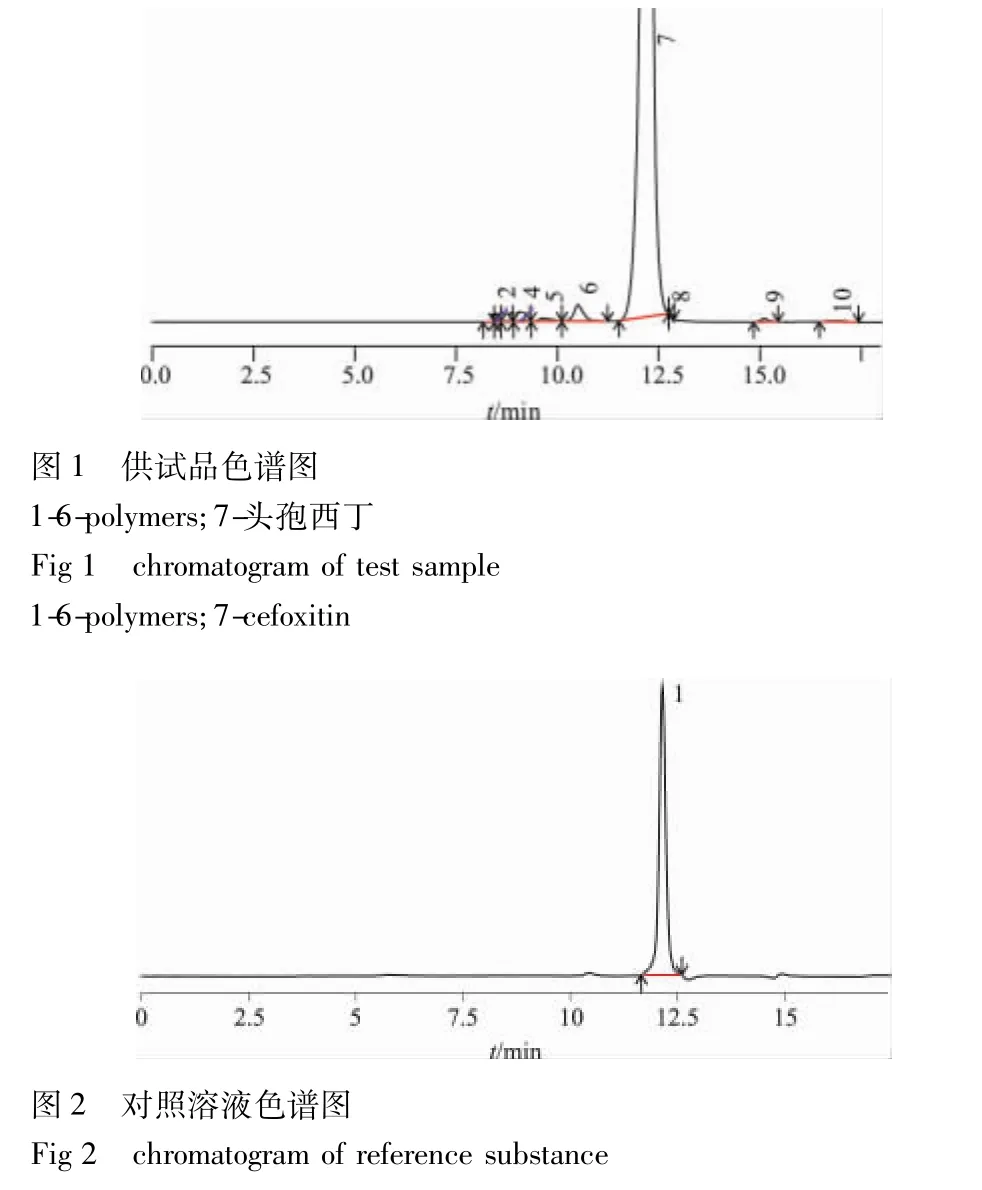
2.3 专属性试验
高温:精密称取对照品约50mg至100mL量瓶中,加水溶解,于100℃水浴中放置30min,取出,冷却到室温,加水稀释至刻度,滤过。
酸破坏:精密称取对照品约50mg至100mL量瓶中,加0.1mol·L-1HCl溶液 10mL,摇匀,静置 4h,加0.1mol·L-1NaOH溶液10mL,用水稀释至刻度,摇匀,滤过。
碱破坏:精密称取对照品约50mg至100mL量瓶中,加0.1mol·L-1NaOH 溶液1mL 溶解,放置5min 后,加0.1mol·L-1HCl溶液1mL,用水稀释至刻度,摇匀,滤过。
氧化:精密称取对照品约50mg至100mL量瓶中,加10%H2O2溶液1mL溶解,放置20min后,用水稀释至刻度,摇匀,滤过。
光照:精密称取对照品约50mg至100mL量瓶中,加水稀释至刻度,摇匀,于5000Lx照度下放置8h,滤过,取续滤液作为供试液。
采用选定的色谱条件和测定方法进样,结果见图3。结果表明:破坏处理之后的样品均有杂质峰出现,其中,热破坏最为严重,碱破坏与酸破坏则次之。采用本法能有效检出产生的降解物,并实现主峰与降解物峰的基线分离。
2.4 检出限及定量限 精密称取对照品适量,用水稀释成每 1mL 中含头孢西丁 5,2.5,1.25,0.5,0.25,0.025 μg 的系列标准溶液。按上述色谱条件,分别取20μL注入液相色谱仪,根据色谱峰以信噪比S/N=3确定头孢西丁最小检出量为0.6ng。以信噪比S/N=10确定头孢西丁的定量限为1.9 ng。
2.5 线性及范围 精密称取对照品适量,用水稀释成每1mL中含头孢西丁 10,5,2.5,1.25,0.5,0.25 μg 的系列标准溶液。各取20μL注入液相色谱仪,记录色谱面积(A)。并以相应组分的色谱峰面积(A)对其浓度(C,μg·mL-1)进行线性回归,得回归方程如下:A=48821C -1728.6,r=0.9999。
结果表明:头孢西丁在0.25 ~10.0 μg·mL-1的浓度范围内线性关系良好。
2.6 精密度 取对照品溶液连续进样5次。头孢西丁峰面积的RSD为0.2%。说明本法精密度较好。
2.7 重复性 平行制备6份供试品溶液,分别立即进样分析,测定聚合物总量,RSD为0.4%,重复性良好。
2.8 稳定性 取含头孢西丁0.5mg·mL-1的样品溶液在室温下放置,于0,1,3.5,6h 分别测定,6h 内杂质峰面积逐渐增大,由此可见,该样品溶液不稳定,不宜长时间放置。
2.9 样品的测定 按本法对3批样品进行检验,测定结果见表1。
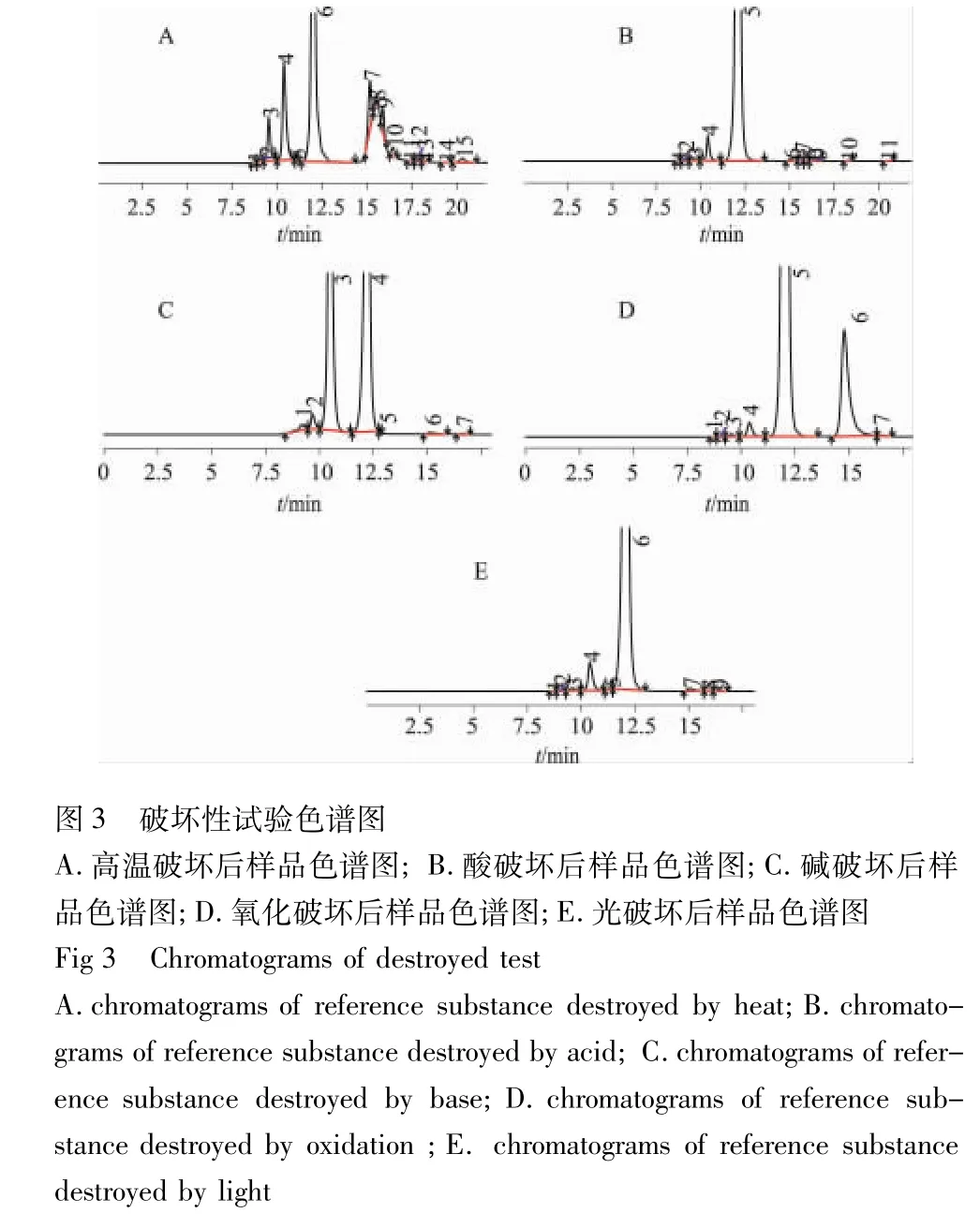

表1 样品聚合物测定结果Tab1 Polymersdeterminationresultsofsamples
3 讨论
(1)缓冲盐的选择 我们对不同离子强度的磷酸盐缓冲液进行了考察,结果如下:A:0.0025mol·L-1磷酸盐缓冲液-乙腈(95∶5)、B:0.005mol·L-1磷酸盐缓冲液-乙腈(95∶5)、C:0.01mol·L-1磷酸盐缓冲液-乙腈(95∶5)、D:0.1mol·L-1磷酸盐缓冲液-乙腈(95∶5)为流动相。色谱图表明,A和D所检出的杂质峰较少且无法实现杂质峰间的分离,B所检出的杂质峰带有肩峰,C共检出6个杂质峰,且实现了主峰与杂质峰及各杂质峰间的有效分离。故选择C作为流动相。
(2)检测波长的选择 用PDA检测器实际测得头孢西丁最大吸收波长为236nm,同一样品中用PDA检测器实际测得的前4个较大杂质峰的紫外吸收峰均为232nm,为保证杂质的最大检出率,选用232nm作为检测波长。
(3)样品中聚合物杂质的控制 由于结构不同的聚合物通常具有相同的生物学特性,且聚合物又具有高度的不均一性,因此,在药品质量控制中只需控制聚合物总量即可[4]。
(4)TSK-GEL 2000SW系球型硅胶基质,在其表面通过共价键化学键合上亲水基团而成,其分离原理综合运用了分子筛效应和凝胶过滤效应,分离能力显著提高,分析时间明显缩短,可以更真实地反映样品聚合物存在状况并避免检测过程中发生新的降解与聚合。但仍有一些细节有待进一步考察与改进。应尽可能将有关物质检查HPLC法与聚合物检查HPLC法检出的各色谱峰逐一鉴定,以避免重复控制。由于β-内酰胺类抗生素在溶液状态下不稳定,分离分析过程中极有可能继续降解与聚合,那么如何制备这些降解物或聚合物并使其保持稳定,是初步测定头孢西丁聚合物的化学结构的关键所在。
[1]尤启东,彭司勋.药物化学[M].北京:化学工业出版社,2004:455-477.
[2]袁雯玮.高分子聚合物研究与中国药典2005年版B-内酰胺类抗生素高分子聚合物修订情况及操作要点[J].中国抗生素杂志,2005,30(12):727-730.
[3]中国药典.二部[S].2010:181.
[4]江晓玲,刘昆,邓俊丰.头孢菌素类抗生素中高分子杂质的研究进展[J].国外医药抗生素分册,2007,28(6):264-269.
[5]顾立素,胡昌勤,金少鸿.安美汀中高分子杂质的分离分析与质量控制[J].药物分析杂志,2001,21(1):13-15.
猜你喜欢
医学食疗与健康(2019年12期)2019-09-10
浙江医学(2018年23期)2018-12-20
——一个解释欧姆表刻度不均匀的好方法
教学考试(高考物理)(2018年6期)2018-12-06
山东化工(2018年15期)2018-09-20
中外医疗(2018年26期)2018-01-05
数学小灵通(1-2年级)(2017年9期)2017-10-13
学苑创造·B版(2017年1期)2017-02-21
小天使·二年级语数英综合(2016年9期)2016-05-14
首都食品与医药(2015年18期)2015-11-03
中国药业(2014年24期)2014-05-26

- 中国医院药学杂志的其它文章
- 高效液相色谱法测定阿托伐他汀钙中的有关物质