
枯草芽孢杆菌启动子的克隆及功能验证
2013-07-25 05:58许丹青李仁宽叶秀云
福州大学学报(自然科学版) 2013年3期
许丹青,李仁宽,2,林 娟,2,严 芬,2,叶秀云,2
(1.福州大学生物科学与工程学院,福建福州 350116;2.酶高效表达国家工程实验室,福建福州 350002)
0 引言
枯草芽孢杆菌是继大肠杆菌后另一重要的模式细菌[1].细菌系统因成本低、发酵周期短和遗传背景清楚等原因成为人们感兴趣的宿主,细菌系统最常用的宿主有大肠杆菌和芽孢杆菌[2].枯草芽孢杆菌是最先应用于表达系统的芽孢杆菌,已被开发成生产蛋白和其他化学产品的生产宿主[3].与大肠杆菌表达系统相比,枯草芽孢杆菌表达系统具有较多的优越性:①无致病性,且被认为是GRAS级的有机体[4];②没有明显的密码子偏好性[5];③可将功能性胞外蛋白分泌到培养基中,简化了分离纯化工艺[6].而大肠杆菌由于其细胞壁结构的原因,在细胞膜外有一层外膜,所以只能将蛋白分泌到周质[7].
自完成基因组测序以来,枯草芽孢杆菌的许多编码基因被鉴定,调节机制和调控元件逐渐被认识.在所有的调控元件中,启动子在枯草芽孢杆菌基因工程菌中扮演着重要角色[8].选择合适的启动子作为调控元件,是重组蛋白高效表达的关键因素之一[9].到目前为止,已经获得一系列的枯草芽孢杆菌启动子,而且有些已经被成功应用于枯草芽孢杆菌表达系统.
本实验室曾从枯草芽孢杆菌中克隆到了可被蔗糖诱导的果聚糖蔗糖酶启动子和信号肽序列PSacB和α-淀粉酶启动子PamyE,在他们的调控下分别实现了枯草芽孢杆菌α-淀粉酶的分泌表达并具有活性.为了分离得到更多的强启动子片段,为枯草芽孢杆菌作为外源蛋白表达宿主的研究奠定基础,本研究构建了以卡那霉素抗性基因为报告基因的启动子探针载体pHT-kan,以鸟枪法处理枯草芽孢杆菌基因组,并在大肠杆菌中分离启动子活性片段,最后在枯草芽孢杆菌中验证启动子的功能.
1 材料和方法
1.1 菌株和质粒
大肠杆菌(E.coli)TOP10、枯草芽孢杆菌(B.subtilis)168和大肠杆菌-枯草杆菌穿梭质粒pHT01[10]均由本实验室保藏.
1.2 酶和试剂
PCR产物纯化回收试剂盒、质粒提取试剂盒和基因组提取试剂盒购自上海生物工程公司;rTaq DNA Polymerase购自TIANGEN公司;限制性内切酶SacⅠ、BamHⅠ、XbaⅠ、Sau3AⅠ、T4 DNA连接酶和低分子量标准蛋白质均购自TaKaRa公司;氨苄青霉素、卡那霉素,山梨醇、甘露醇、琼脂糖、酵母提取物、蛋白胨和氯化钠等购自上海生物工程公司.
1.3 培养基
大肠杆菌、枯草芽孢杆菌的培养采用LB培养基,根据实验需要添加适当浓度的抗生素,氨苄青霉素(AMP)的终浓度为 100 μg·mL-1(E.coli);卡那霉素(KM)的终浓度为 40 μg·mL-1(E.coli)和 10 μg·mL-1(B.subtilis).
1.4 枯草芽孢杆菌基因组DNA的提取
取-80℃保藏的枯草芽孢杆菌168菌株,接种于5 mL LB培养基,37℃,200 r·min-1过夜培养.取1 mL过夜培养物12 000 r·min-1离心1 min,然后采用Ezup柱式基因组DNA抽提试剂盒(细菌)提取基因组DNA.
1.5 卡那霉素抗性基因的克隆及启动子探针的构建
根据pET28a(+)中卡那霉素抗性基因的序列,利用VectorNTI软件设计合成一对引物P1、P2,用于克隆不含启动子的卡那霉素抗性基因.

其中:P1为卡那霉素抗性基因上游引物,在起始密码子ATG的上游在SacⅠ后引入BamHⅠ(GGATCC)酶切位点,用于启动子片段的插入.以含有卡那霉素抗性基因的质粒pET28a(+)为模板,扩增卡那霉素抗性基因片段.纯化回收的PCR产物与PCR2.1克隆载体4℃连接过夜,转入大肠杆菌TOP10感受态细胞,经菌落PCR验证后,提取质粒酶切验证,构建成功的载体命名为PCR2.1-kan.
PCR2.1-kan用SacⅠ、XbaⅠ双酶切凝胶纯化回收小片段约816 bp,pHT01经同样双酶切纯化回收大片段,两种纯化回收产物连接过夜后转化大肠杆菌TOP10感受态细胞,经菌落PCR验证和酶切验证,送测.构建成功的启动子探针命名为pHT-kan.
1.6 启动子活性片段的克隆
采用同尾酶的策略,BamHⅠ(识别序列为GGATCC)与Sau3AⅠ(识别序列为GATC)的粘性末端相同,且Sau3AⅠ识别4个碱基,因此,理论上枯草芽孢杆菌基因组可被Sau3AⅠ切成256 bp左右的片段,这与枯草芽孢杆菌启动子片段大小100~500 bp左右相接近.用Sau3AⅠ酶切枯草芽孢杆菌168基因组,纯化回收后与经BamHⅠ酶切并去磷酸化的pHT-kan连接,转化大肠杆菌TOP10感受态细胞,涂布于40 μg·mL-1卡那霉素抗性LB平板.
从卡那霉素抗性平板中挑取长的大而圆的转化子,点种在100 μg·mL-1卡那霉素平板上.用含不同卡那霉素浓度梯度的LB液体培养基检测所有启动子的抗性水平.最后筛选出抗性最高的几个启动子,对其进行菌落PCR验证并测序.
1.7 启动子测序及序列的分析
在SacⅠ上游设计合成测序引物P3:5’-GCTGCAAGGCGATTAAGTTGGGTAACGCC-3’,对抗性最高的几个启动子进行测序和序列分析.通过以下在线分析网站分析启动子片段:
http://www.ncbi.nlm.nih.gov/ NCBI;
http://www.fruitfly.org/ 启动子在线分析网站;
http://genolist.pasteur.fr/SubtiList/ 枯草芽孢杆菌168全基因组数据库.
对测序的结果进行分析,根据预测的启动子功能区域,结合枯草芽孢杆菌σ因子的特点,推测其-35区、-10区,根据枯草芽孢杆菌数据库确定核糖体结合位点RBS,选取其中的几个启动子进行启动子功能验证.
1.8 启动子功能验证
选取几个经测序和序列分析的重组子提取质粒,采用电转法[11]转入枯草芽孢杆菌168感受态细胞,重组菌经含卡那霉素抗性LB平板筛选后采用卡那霉素抗性梯度验证启动子的强度.
2 结果
2.1 枯草芽孢杆菌基因组DNA的提取
采用Ezup柱式基因组DNA抽提试剂盒(细菌)提取基因组DNA,经1%琼脂糖凝胶电泳验证提取成功后方可进行后续试验.
2.2 卡那霉素抗性基因的克隆及启动子探针的构建
采用设计的引物P1、P2,以pET28a(+)为模板,通过PCR扩增出卡那霉素抗性基因.TA克隆连接到PCR2.1上,构建PCR2.1-kan.PCR2.1-kan经SacⅠ、XbaⅠ双酶切后,被切成两条带,结果见图1.说明目的基因已经克隆到PCR2.1上.
pHT01-kan经SacⅠ、XbaⅠ双酶切后,被切成两条带,证明目的基因已经插入pHT01(图2),启动子探针载体构建成功.
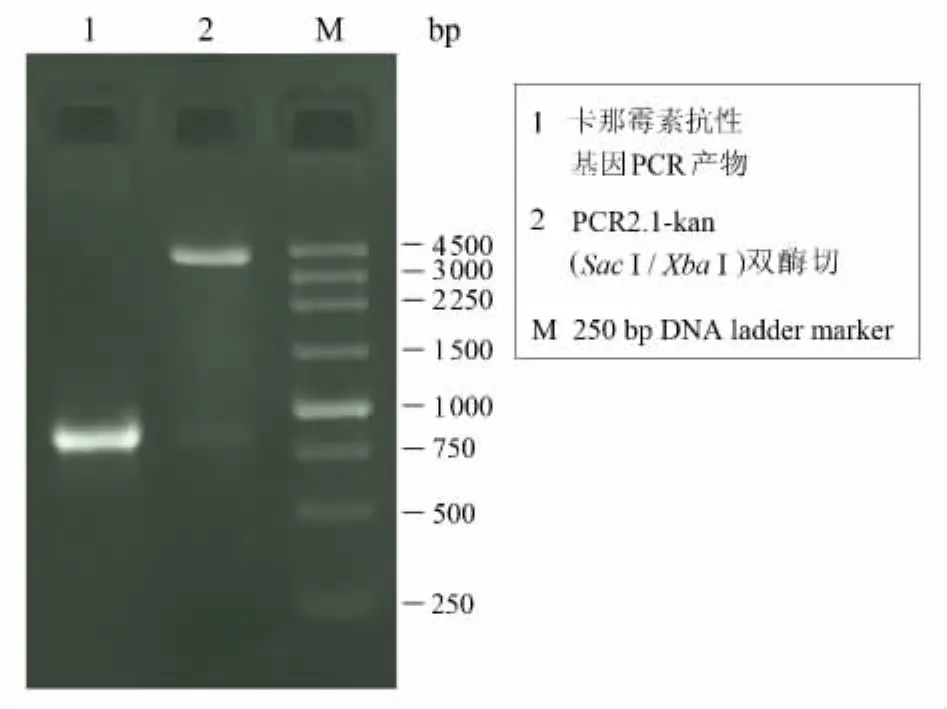
图1 PCR2.1-kan双酶切验证Fig.1 Identification of PCR2.1 - kan by enzyme digestion
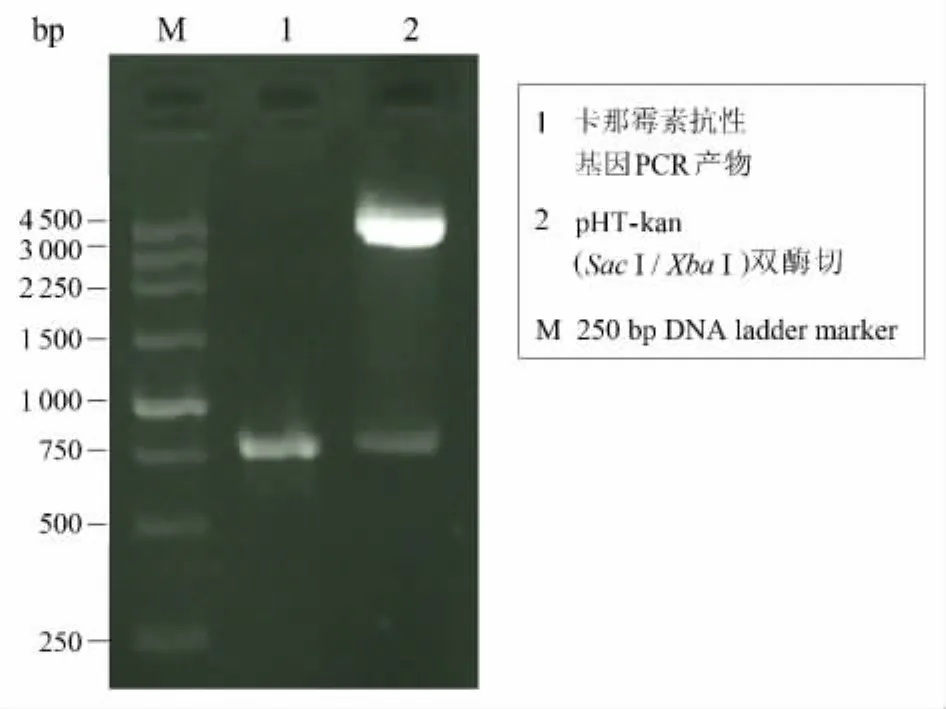
图2 pHT-kan双酶切验证Fig.2 Identification of pHT - kan by enzyme digestion
2.3 启动子活性片段的克隆
取适量的枯草芽孢杆菌基因组DNA在37℃进行酶切,控制酶切反应体系中的Sau3AⅠ酶活单位,使得基因组DNA经酶切1 h后的片段集中在100~1 000 bp之间,pHT-kan经BamHⅠ酶切并去磷酸化,纯化回收上述酶切产物,结果见图3.
取上述回收产物各适量酶连,转化大肠杆菌TOP感受态细胞,涂布于40 μg·mL-1卡那霉素抗性LB平板.经验证几乎所有具有抗性的转化子均可在100 μg·mL-1的卡那霉素平板上生长.挑取100个大而圆转化子,点种在100 μg·mL-1卡那霉素抗性LB平板上保存,依次命名为pHT-BSPx-kan,x=1,2,…,10.将这些转化子接种在500、1 000、2 000和3 000 μg·mL-1卡那霉素液体LB中进行抗性梯度筛选.实验表明随着卡那霉素浓度的升高,转化子的生长个数逐渐减少,因此根据转化子的生长水平,可反应启动子活性片段在大肠杆菌中对卡那霉素抗性基因的启动强度.最终获得高抗性转化子pHT-BSP25-kan和 pHT -BSP31 -kan,他们的抗性水平可达2 000 μg·mL-1.
对其中抗性较高的启动子进行菌落PCR验证,验证所采用的引物在卡那霉素抗性基因插入位点的上游和下游.若启动子探针未插入启动子活性片段,则PCR产物大小在1 000 bp左右,由图4可以看出:PCR产物菌落在1 000~1 500 bp之间,因此启动子活性片段小于500 bp,这与枯草芽孢杆菌启动子片段大小相符.
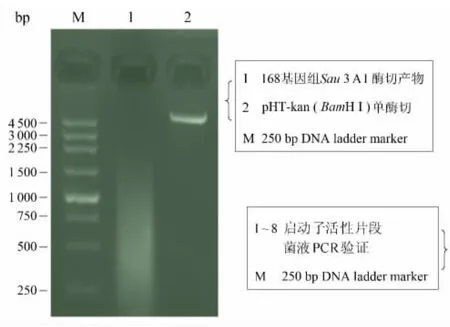
图3 启动子片段的克隆Fig.3 Cloning of promoter fragment
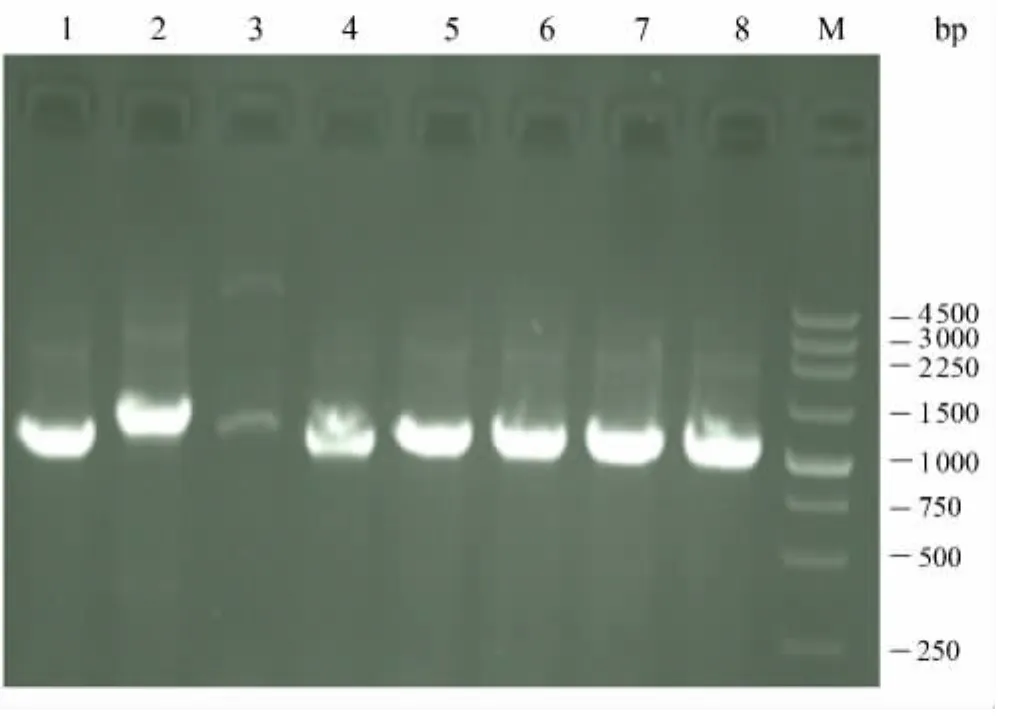
图4 启动子活性克隆菌落PCR验证Fig.4 PCR analysis of recombinant clones
2.4 启动子测序及序列分析
对启动子活性片段BSP25和BSP31进行测序,序列测定由上海生工完成.序列如图5.

图5 启动子序列分析Fig.5 Analysis of promoters
BSP25长242 bp,经Blast序列比对,与枯草芽孢杆菌168基因组同源性达100%,查询枯草芽孢杆菌168基因组数据库,前133 bp在yhjO的3’端,从131个碱基开始的3个碱基TAG为yhjO的终止子.yhjN基因的启动子部分与yhjO的3’端重叠,这与原核细菌基因的重叠现象一致,最后3个碱基为yhjN基因的起始密码子TTG,起始密码子后有10个氨基酸与卡那霉素抗性基因发生了融合,但后者的阅读框并没有改变,因此不影响卡那霉素抗性基因的活性.
BSP31长128 bp,经Blast序列比对,与枯草芽孢杆菌168基因组同源性达99%,查询枯草芽孢杆菌168基因组数据库,它是yktD基因的启动子,其第一个碱基再往上游228 bp处为nprE的终止子TTG,最后3个碱基为起始密码子ATG.yhjN和yktD基因的功能未知.
采用启动子在线分析网站对BSP25和BSP31进行启动子功能区域预测(表1),结果表明两者均含有启动子功能片段,启动子可能性的预测值均大于94%.

表1 启动子BSP25、BSP31结构预测Tab.1 Promoter prediction for BSP25 and BSP31
根据预测的结果,结合枯草芽孢杆菌σ因子的特点,预测启动子区域的-35区、-10区,结果标在序列处.其中BSP25与营养生长时期σC识别的启动子相似,BSP31与早期生长时期σA识别的启动子相似.核糖体结合位点RBS的序列则由枯草芽孢杆菌基因组数据库直接确定.
2.5 启动子功能验证
提取质粒,采用电转法将含有BSP25和BSP31片段的质粒pHT-BSP25-kan和pHT-BSP31-kan转入枯草芽孢杆菌168感受态细胞,涂布卡那霉素抗性平板,过夜培养后长出具有卡那霉素抗性的转化子.实验结果表明,启动子BSP25和BSP31能够启动卡那霉素在枯草芽孢杆菌中表达.挑取其中的几个克隆接种在含500、1 000、2 000和3 000 μg·mL-1卡那霉素LB液体培养基中,检查其抗性水平,实验结果列于表2.
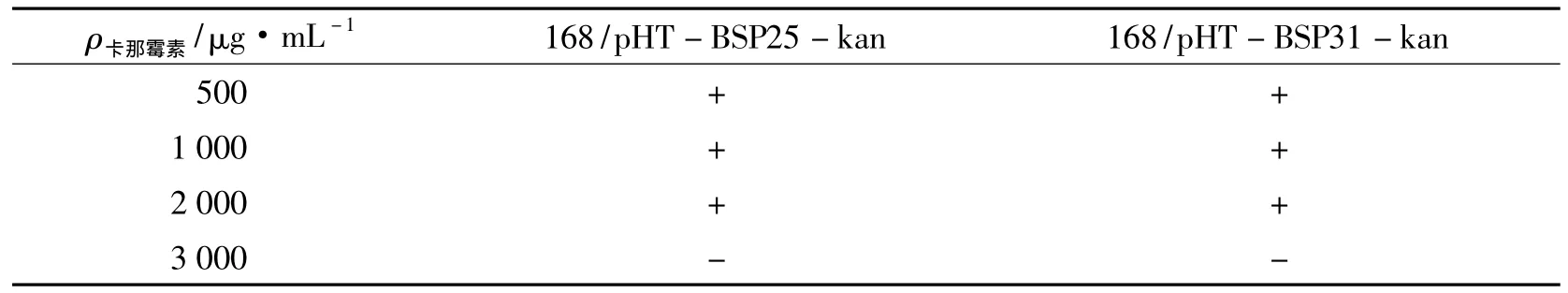
表2 重组枯草芽孢杆菌的卡那霉素抗性水平Tab.2 Kanamycin resistance level of the recombinant B.subtilis
实验表明,168/pHT-BSP25-kan和168/pHT-BSP31-kan均可在2 000 μg·mL-1卡那霉素液体LB培养基中生长,其抗性水平和在大肠杆菌中相当.该实验结果表明在大肠杆菌中克隆筛选得到的启动子片段,可在枯草芽孢杆菌中启动卡那霉素抗性基因的表达.因此采用在大肠杆菌中筛选枯草芽孢杆菌启动子的方法是可行的.
3 讨论
尽管枯草芽孢杆菌168已于1997年完成全基因组测序,且一些公认的启动子如PSacB[12]、P43[13]、PamyE[14]等已被较多地研究和应用.但到目前为止,枯草芽孢杆菌基因组的很多基因功能尚属未知.因此,有必要继续克隆分析得到更多的强启动子活性片段,为枯草芽孢杆菌表达系统的应用奠定基础.
启动子克隆的常用方法有启动子探针载体、环状PCR和TAlL-PCR等,其中枯草芽孢杆菌最常用的启动子克隆方法是启动子探针载体.构建启动子探针载体时常用的报告基因有:绿色荧光蛋白基因和抗生素抗性基因,还有一些实验室利用特定的酶作为报告基因.以绿色荧光蛋白作为报到基因还需要通过荧光光度计来测定才能分析启动子强度,仪器设备较为繁琐.pHT01本身具有氨苄青霉素和氯霉素抗性,因此本实验采用卡那霉素抗性基因作为报告基因,且用卡那霉素浓度梯度筛选,操作简单,并能较好地指示启动子的强度.
pHT01载体是MoBiTec公司的大肠杆菌-枯草芽孢杆菌穿梭质粒,因此在大肠杆菌和枯草芽孢杆菌中均有自主复制的功能,如果启动子适合也可在两种宿主菌中实现目的基因的表达.用不含启动子的卡那霉素抗性基因替换其启动子和LacI部分,故可用于筛选具有启动子活性的枯草芽孢杆菌碱基片段.实验研究表明,在大肠杆菌中筛选的启动子活性片段也能在枯草芽孢杆菌中调控目的基因的表达,因此,该启动子克隆方法可为枯草芽孢杆菌表达系统的研究提供实验支持.
构建启动子筛选载体,建立一种枯草芽孢杆菌启动子筛选和启动子活性验证的方法.对于筛选到的较高效启动子,今后可开展对启动子片段进行删减实验,或者把BSP25起始密码子后10个氨基酸去掉,进一步研究启动子活性的变化情况.
[1]郝彤.枯草芽孢杆菌高质量代谢网络的初步构建[D].天津:天津大学,2007.
[2]Terpe K.Overview of bacterial expression systems for heterologous protein production:from molecular and biochemical fundamentals to commercial systems[J].Appl Microbiol Biotechnol,2006,72:211 -222.
[3]Park Y C,Jun S Y,Seo J H.Construction and characterization of recombinantBacillus subtilisJY123 able to transport xylose efficiently[J].Journal of Biotechnology,2012,161:402 -406.
[4]夏雨.枯草芽孢杆菌食品级表达系统的构建和分泌表达研究[D].无锡:江南大学,2007.
[5]Vavrova L,Muchova K,Barak I.Comparison of differentBacillus subtilisexpression systems[J].Research in Microbiology,2010,161:791-797.
[6]Simonen M,Palva I.Protein secretion inBacillusspecies[J].Microbiological Reviews,1993,57(1):109 -137.
[7]周冰,张惟材.枯草芽孢杆菌蛋白质分泌机制研究进展[J].生物技术通讯,2004,15(3):281-284.
[8]Zhang Ai- ling,Liu Hui,Yang Ming - ming,et al.Assay and characterization of a strong promoter element fromB.subtilis[J].Biochemical and Biophysical Research Communications,2007,354:90 -95.
[9]佟小雪,牛丹丹,陈献忠,等.地衣芽孢杆菌高温α-淀粉酶异源表达研究[J].生物技术,2009,19(5):11-13.
[10]Phan T T P,Nguyen H D,Schumann W.Novel plasmid-based expression vectors for intra- and extracellular production of recombinant proteins inBacillus subtilis[J].Protein Expression and Purification,2006,46:189 -195.
[11]Xue Gang-ping,Johnson J S,Dalrymple B P.High osmolarity improves the electro-transformation efficiency of the gram -positive bacteriaBacillus subtilisandBacillus licheniformis[J].Journal of Microbiological Methods,1999,34:183 -191.
[12]Liu Sen-lin,Du Kun.Enhanced expression of an endoglucanase inBacillus subtilisby using the sucrose-induciblesacB promoter and improved properties of the recombinant enzyme[J].Protein Expression and Purification,2012,83:164 -168.
[13]Contreras J A R,Pedraza-Reyes M,Ordoñez L G,et al.Replicative and integrative plasmids for production of human interferon gamma inBacillus subtilis[J].Plasmid,2010,64:170 -176.
[14]Kim J H,Hwang B Y,Roh J,et al.Comparison of PaprE,PamyE,and PP43promoter strength for β - galactosidase and staphylokinase expression inBacillus subtilis[J].Biotechnology and Bioprocess Engineering,2008,3:313 -318.
猜你喜欢
湖南饲料(2021年4期)2021-10-13
西南农业学报(2021年4期)2021-05-25
农药科学与管理(2019年6期)2019-11-23
农药科学与管理(2019年6期)2019-11-23
农药科学与管理(2019年8期)2019-11-23
扬子江(2019年3期)2019-05-24
山西农业大学学报(自然科学版)(2017年9期)2017-08-09
当代化工研究(2016年7期)2016-03-20
分析测试学报(2015年5期)2016-01-13
江苏大学学报(医学版)(2015年5期)2015-04-16
