
锂离子电池聚合物正极材料研究进展
2013-07-18 07:09:46王运灿郝建原
化工进展 2013年1期
王运灿,罗 琳,刘 钰,郝建原,2
(1电子科技大学微电子与固体电子学院,四川 成都 610054;2电子科技大学电子薄膜与集成器件国家重点实验室,四川 成都 610054)
随着人类社会的快速发展以及传统天然矿物资源的日趋枯竭,能源和环境危机已然成为全球社会亟待解决的首要问题。锂离子电池作为电化学能源发展的最新技术领域,具有开路电压高、比能量大、循环寿命长、无记忆效应、无污染以及自放电率小等各项优异性能,在移动通讯、笔记本电脑、数字处理机等便携式电子产品以及电动汽车的发展和研究中,都起着不可替代的作用[1-3]。
目前商用化的锂电池正极大多采用 LiCoO2、LiNiO2、LiMn2O4等无机锂盐材料,虽然这些材料放电容量、可逆性、充放电效率等性能表现稳定良好,但是也存在着矿物资源昂贵、有毒,正极材料容量有限等不利因素,严重制约了锂离子电池在电源行业的发展和生产[4-6]。
聚合物锂离子电池使用高分子聚合物替代传统锂离子电池使用的无机锂盐作正极,让锂离子电池的研究进入了全新的发展阶段。相比于传统的能源电池,聚合物锂离子电池具有更高的能量密度、电池尺寸更小型化、更安全更环保等诸多优点,成为了全球锂电行业研究的新浪潮。目前研究开发的聚合物正极材料主要有4类:自由基聚合物正极材料、导电聚合物正极材料、有机多硫聚合物正极材料以及多骨架碳硫交联聚合物正极材料。
1 自由基聚合物正极材料
1.1 自由基聚合物的结构
自1956年发现稳定氮氧自由基以来[7],自由基化合物在生物化学领域的研究和应用引起了人们的广泛兴趣。自由基聚合物一般是指侧链带有稳定氮氧自由基基团的一类聚合物高分子,其中研究最多和发展前景最好的是 2,2,6,6-四甲基-4-羟基哌啶-1-氧自由基(2,2,6,6-tetramethyl-4-piperidine,简称TEMPO)及其衍生物。TEMPO的分子结构如图1所示,分子结构中的甲基官能团非常稳定,与大多数试剂都不起反应,因而被广泛用于癌化学、辐射化学、激发态化学以及自旋标记技术中[8-14]。

图1 TEMPO结构示意图
为了更好研究氮氧自由基的反应机理,Linnett等[15]、Kawamura 等[16]、Hoffman 等[17]分别从非配对空间轨道和休克尔分子轨道出发,提出了氮氧自由基具有异核双原子三电子键的结构,三电子键在不同情况下有3种可能的结构,如图2所示。

图2 氮氧自由基三电子结构
结构Ⅲ是中性自由基,一般都用它表示氮氧自由基的电子构型。氮氧自由基的自旋密度主要集中在N和O上,而且几乎全在2pz轨道上,表现出稳定的顺磁性[18]。正是由于这种电子结构,氮氧自由基不会像其它自由基一样发生二聚化反应,在加热、蒸馏等反应中也表现异常稳定。
1.2 氮氧自由基的制备
TEMPO及其衍生物通常以2,2,6,6-四甲基哌啶为前体,经过氧化剂(如过氧化氢或叔丁基过氧化氢)氧化侧链的亚胺基团而获得。另外,在反应体系中加入钨酸钠、间氯过氧苯甲酸作为催化剂,经均相催化生成稳定的氮氧自由基[19-22]。
1.3 自由基聚合物正极材料
锂离子电池正极材料使用自由基聚合物的研究始于近几年,氮氧电子结构在充放电循环过程中,反复发生氧化还原反应而自由基分子不断裂,且不产生单个的阴离子和阳离子自由基,表现出优良的稳定性能和快速充放电性能。充电时•O—N基团失去电子被氧化为 O=N⊕,放电时 O=N⊕又通过得到电子而还原为•O—N。
日本的 Nakahara等[23]和我国中南大学邓凌峰课题组[24],分别用不同的方法合成出了PTMA自由基聚合物。充放电性能结果显示出78 mA·h/g的容量,完成500次的充放电循环后,依然保持着90%以上的放电容量;循环伏安曲线显示,氧化峰与还原峰之间的点位差仅为63 mV,说明正极材料发生氧化还原的速度很快,表现出高的功率密度和快速充放电性能。
刘承梅等[25]利用具有高导电率和高比表面积的碳黑Black Pearl-2000(BP)研制了PTMA-BP电极,并深入研究了PTMA含量和电极厚度对材料比容量、充放电效率和循环性能的影响。结果表明,厚度为20 μm,PTMA含量为22.5%时电极具有151 mA·h/g的最高比容量和最优的循环性能,500次充放电循环后的容量保持率大于88%。并且倍率性能优异,以50 C充放电,电极依然显示130 mA·h/g的比容量。
2 导电聚合物正极材料
导电聚合物的发现源于 1977年, Heeger、MacDiarmid和白川英树等[26]在聚乙炔薄膜中掺杂碘后其导电性从10−6S/cm增加到103S/cm,从此改变了有机聚合物都是绝缘体的传统认识,兴起了导电聚合物在电池行业的研究热潮。
2.1 导电聚合物的结构
导电聚合物通常是分子结构中存在着共轭π键和孤对电子,这类分子经化学或电化学掺杂后,会表现出很高的电导率。常见的几类导电聚合物有聚乙炔(PA)、聚苯胺(PAn)、聚吡咯(PPy)、聚对苯(PPP),表1列出了它们的分子结构及电导率。
2.2 导电聚合物的掺杂及导电机理
研究发现,纯净的共轭聚合物本征态,由于其结构链的规整度,电子难以在各π轨道间顺利跃迁,其导电率很低。通过掺杂,向高分子链中引入电子(还原)或者移走高分子链中的电子(氧化),从而改变π电子能带的能级,聚合物的导电性就会大大增加。目前,常用的掺杂方法有化学掺杂和电化学掺杂。
(1)化学掺杂 共轭聚合物与氧化剂或还原剂之间发生得失电子的氧化还原反应,从而改变链结构上的电子离域分布。根据氧化还原的机理,化学掺杂又可分为P型掺杂和N型掺杂。
P型掺杂见式(1)。

式中,CP为共轭聚合物。
另外还有一种化学掺杂的方法叫质子酸掺杂,在质子酸的掺杂机制中,共轭聚合物与掺杂剂之间并无明显的电荷转移,质子以黏附的形式附加在聚合物链上,所带的电荷分散在链中,同时质子酸中的阴离子成为对阴离子。可溶性导电聚苯胺通常采用的就是质子酸掺杂,其掺杂前后的电子结构分布如图3所示。

图3 本征态PAn与掺杂态Pan
(2)电化学掺杂 许多共轭聚合物在高电位区能够发生电化学P型掺杂/脱掺杂过程,在低电位区又可发生电化学N型掺杂/脱掺杂过程。因此,利用电化学反应,也可以实现导电聚合物的掺杂[27]。发生电化学P型掺杂时,共轭链被氧化,其价带失去电子并伴随着对阴离子的掺杂;相反,发生电化学N型掺杂时,共轭链被还原,价带得到电子并伴随对阳离子的掺杂。其掺杂过程如式样(3)、式(4)所示。
P型掺杂见式(3)。
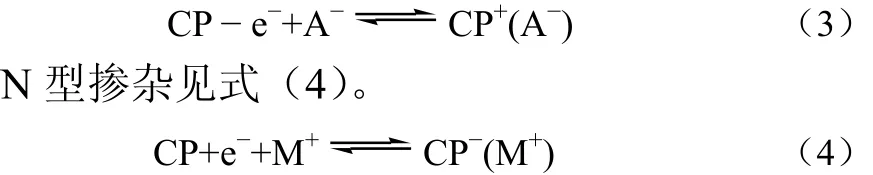
2.3 导电聚合物的制备及在锂电池正极材料中的应用
不同的导电聚合物需要不同的聚合方法,比如聚乙炔需要特殊的催化剂通过化学合成来制备[28-29]。也有一些通用的方法,能够适用于各种不同的导电聚合物的制备,例如通过单体氧化聚合或电化学氧化聚合的方法可以制备聚吡咯、聚苯胺、聚噻吩和各种杂环聚合物。

表1 导电聚合物分子结构及电导率
(1)化学氧化合成 使用三氯化铁、过硫酸铵等氧化剂在溶剂中将单体氧化聚合。例如Chaing等[30]用过硫酸铵在盐酸介质中氧化苯胺单体,得到经盐酸掺杂后的导电聚苯胺(Pan-HCl)粉末,之后经过氨水脱掺杂、NMP溶解后再浇铸,制得了导电聚苯胺膜。
(2)电化学聚合 聚吡咯、聚苯胺、聚噻吩等都可以通过电化学聚合其单体来制备。不同的导电聚合物单体有不同的聚合电位,聚合电位越高,单体在电解质中聚合越困难,聚合电位越低,则聚合反应进行的就相对容易。表2为几种导电聚合物单体的聚合电位。
吡咯单体的聚合电位只有0.7 Vvs.SCE,有机电解液和水盐溶液都可以作为电解聚合的电解液。有机电解液中,聚合物单体的溶解度高,反应单一,不会存在溶剂副反应。常用的有机电解液有乙腈、碳酸丙烯酯(PC)等,另外使用高氯酸锂、对甲苯磺酸作为支持电解质。使用水盐溶液作电解液时,溶液pH值、支持电解质的阴离子对吡咯单体的聚合会有很大影响,弱酸性(2 < pH < 5)、表面活性阴离子电解液中制备的导电聚吡咯膜电导率较高。自从导电聚合物的合成工艺被成熟开发以来,其在电极材料领域的应用也广泛发展。Chen Show-An等[31-32]采用在聚苯胺电极中掺加锂盐的方法,使聚苯胺材料达到了221 W·h/kg的能量密度和91%的平均库仑效率。Genies等[33]用电化学方法制备的聚苯胺自支撑膜,组装了钮扣式和卷绕式的二次模型电池,以0.25 mA/cm2放电,其比能量和比容量分别为400 W·h/kg和130 mA·h/g,充放电可达几百次以上。
3 有机多硫聚合物正极材料
单质硫的理论比容量是 1675 mA·h/g,与锂组装成电池的理论比能量可达2600 W·h/kg,且矿物硫来源丰富、廉价易得,是很有价值的锂电池正极材料替代品。但是单质硫与锂组成的电池体系,由于硫本身的绝缘性,且电极反应产生的中间产物Li2Sx易于溶解在电解液和沉积在锂负极表面,严重影响了电池的充放电功率和循环性能[34]。

表2 几种单体的氧化聚合电位
有机多硫聚合物正极材料将 S—S键引入有机物分子中,形成各种线性、梯形或者网状多交联的硫化聚合物。
3.1 线性多硫聚合物
20世纪 90年代初,Liu等[35]发现 2,5-二巯基-1,3,4-噻二唑(DMcT)单体经聚合得到的线性聚有机二硫化物PDMct(图4),用于锂电池正极材料具有很高的理论比容量(357 mA·h/g)。但是由于DMcT本身的导电性很差,故通常将导电聚合物或其它活性物质与其复合改性,Naoi等[36]用聚苯胺PAn对DMcT进行复合,发现氧化还原电流增大,阴极还原峰正移,证明了PAn对DMcT有催化作用。又有研究发现,在DMcT/PAn 的复合电极上,DMcT是主要的活性物质,PAn 既是催化剂,又有电化学反应活性,并且在分子水平上起到集流体的作用[37]。
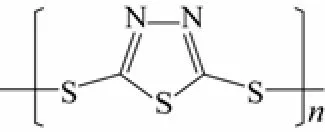
图4 PDMcT的化学结构式
3.2 梯形多硫聚合物
基于线性多硫聚合物复合导电聚合物的思想,Naoi等[38]又设想将两种结构合成到一种活性聚合物中,形成新的兼有两种结构优点的新型正极材料,Naoi用电化学聚合得到梯形结构的聚(2,2'-二硫代)二苯胺(PDTDA)(图5)。这种结构的分子,活性S—S键被连接到聚苯胺链结构之间,氧化还原反应形成的—S−Li+等小分子,始终被聚苯胺长链结构束缚,保证了电极材料良好的反应活性和循环性能。Uemachi等[39]根据同样的思想合成了 1,4-亚苯基-1,2,4-二噻唑聚合物(PPDTA)(图6),分子中兼有S—S键和7-π-电子不饱和1,2,4-二噻唑啉环。电化学性能测试放电容量达到420 mA·h/g,是常规锂离子电池正极材料的3~4倍。
3.3 网状多交联硫化聚合物

图6 PPDTA的化学结构式
1999年,Gorkovenko等[40-41]报道了一类新型的网状多交联硫化聚合物正极材料,他们以六氯环戊二烯、六氯代苯等环状多卤代烃为原料,与多硫化钠反应,制备了一系列网状多硫代聚合物(图7)。这类材料结构中心包含3~12元碳环,环之间通过—(S—S)n—多硫键链接,放电容量高达 1000 mA·h/g以上,是一类很有前途的高比容量电池材料。但是由于放电结束后,聚合物终将解聚为小分子单体,故仍未满足实用化的循环要求,仅仅只能用于一次电池体系。

图7 网状多交联硫化聚合物
4 多骨架碳硫交联聚合物正极材料
在第十一届全国电化学会议上,杨裕生院士[42]提出了“主链导电,侧链储能”的电极材料设计思路,这类聚合物主链为导电高分子,侧链为可发生氧化还原反应的多硫键,材料本身有着很高的硫碳比和导电性,因此理论比容量很高。
李晓林等[43]根据这个思路,用偏二氯乙烯制备的碳炔与升华硫以摩尔比1∶5混合球磨,制备的产物经13CNMR、Raman等表征,显示出多硫化碳炔的结构特性,之后经电化学性能测试,首次放电容量达 600 mA·h/g,充放电效率接近 100%。Zhang Shichao等[44]也用聚吡咯经盐酸、过氧化氢反应先制取氯代聚吡咯,之后与多硫化钠反应,获得了多硫代聚吡咯正极材料,材料首次放电容量达 515 mA·h/g,20次充放电循环后也保持着 452 mA·h/g的放电容量。
5 结 语
传统的无机锂盐正极材料,诸如层状钴酸锂、尖晶石锰酸锂、橄榄石磷酸锂铁等,虽然在充放电效率和可逆性等性能上表现优异,但比容量较低,平均约为150 mA·h/g,且存在有其它弊端和局限性。例如钴金属价格昂贵,成本过高;锰酸锂受 Jahn-Teller效应的影响,其结构不太稳定,循环性能较差,实际容量也偏低。新型的聚合物正极材料包含了自由基聚合物、导电聚合物、有机多硫聚合物以及多骨架碳硫交联聚合物等,它们的比容量能达到几百甚至上千,原料廉价易得,是未来电动汽车广泛发展的潜在正极材料。但是,目前聚合物正极材料还存在着容量衰减快、制备工艺不成熟以及易降解等问题,有待人们进一步地研究和解决。如果能够找出克服上述不利因素的方法,相信会是人类能源和科学界一个不朽的进步。
[1]Bruno Scrosati,Jürgen Garche.Lithium batteries:Status,prospects and future[J].Journal of Power Sources,2010,195:2419-2430.
[2]Liu Chang,Li Feng,Ma Laipeng,et al.Advanced materials for energy storage[J].Adv.Mater.,2010,22:E28-E62.
[3]陈猛,史鹏飞,程新群.塑料锂离子电池研究概况[J].电池,2000,30(3):129-133.
[4]Jeffrey W Fergus.Recent developments in cathode materials for lithium ion batteries[J].Journal of Power Sources,2010,195:939-954
[5]Boone B Owens.Solid state electrolytes:Overview of materials and applications during the last third of the Twentieth Century[J].Journal of Power Sources,2000,90:2-8.
[6]Schoonman J,Tuller H L,Kelder E M.Defect chemical aspects of lithium-ion battery cathodes[J].Journal of Power Sources,1999,81-82:44-48.
[7]Johnson D,Rogesr M,Trappe G.Aliphatic hydroxylamines(Ⅱ):Autoxidation [J].J.Chem.Soc.,1956,10:1093-1095.
[8]Dvais T D,Christoffersen R E,Maggiora G M.Ab initio calculation on large moleucles using molecular fragments.Nitroxide spin label characterization[J].Journal of the American Chemical Society,1975,97(6):1347-1354.
[9]Fuchs J,Groth N,Herrling T,et al.Electron paramagnetic resonance studies on nitroxide radical 2,2,5,5–tetramethyl-4- piperidin-1-oxyl(TEMPO) redox reactions in human skin[J].Free Radical Biology and Medicine,1997,22(6):967- 976.
[10]John W M,Michael F C,Kim B M,et al.Nitroxide mediated living radical polymerazation of styrene in miniemulsion:Modelling persulfate-initiated systems[J].Chemical Engineering Science,2003,58(7):1177-1190.
[11]Magin I M,Shevel’kov V S,Obynochny A A,et al.CIDNP study of the third spin effect on the singlet-triplet evolution in radical pairs[J].Chemical Physics Letters,2002,357(5-6):351-357.
[12]Fujiwara H,Fujiwara E,Kobayashi H.Synthesis,structures and properties of new of organic donors connecting to a TEMPO radical through a pyrrolidine ring[J].Synthetic Metals,2003,133-134(13):359-360.
[13]Mohler D L,Dain D R,Kerekes A D,et al.Organometallic photonucleases:A novel class of DNA-cleaving agents[J].Bioorganic & Medicinal Chemistry Letters,1998,8(7):871-874.
[14]Damiani E,Greci L,Parsons R,et al.Nitroxide radicals protect DNA from damage when illuminated in vitro in the presence of dibenzoylmethane and a common sunscreen ingredient[J].Free Radical Biology and Medicine,1999,26(7-8):809-816.
[15]Linnett J W,Rosenberg R M.Structure and properties of nitroso compounds[J].Tetrahedron,1964,20(1):53-66.
[16]Kawamurra T,Matsunami S,Yonezawa T.Solvent effect on theg-value of di-t-butyl nitric oxide[J].Bull.Chem.Soc.Japan,1967,40(5):1111-1115.
[17]Cohen A H,Hoffman B M.Nitrogen-14 and oxygen-17 hyperfine interactions in perturbed nitroxides[J].J.Phys.Chem.,1974,78(13):1313-1321.
[18]刘有成,江致勤,吴树屏.氮氧自由基的研究(Ⅱ)-哌啶类氮氧自由基的溶剂效应及其分子结构的研究[J].高等学校化学学报,1980,1(2):67-73.
[19]Elmer J,Gerald M,Mohaamed B,et al.Improved methods for the oxidation of secondary amines to nitroxides[J].Synthhetic Communications,1975,5(6):409-412.
[20]Neiman M B,Rozanzev E G,Mameodova Y G.Free radical reactions involving nounpaired electrons[J].Nature,1962,196:472-474.
[21]Rozanzev E G,Neiman M B.Organic radical reactions involving no free valence[J].Tetrahedron,1964,20:131-137.
[22]Rauckman E J,Rosen G M,Aboou Donia M B.Improved methods for the oxidation of secondary amines to nitroxides[J].Synthetic Communications,1975,5:409-413.
[23]Nakahara K,Iwasa S,Satoh M,et a1.Rechargeable batteries with organic radical cathodes[J].Chemical Physics Letters,2002,359(5-6):351-354.
[24]邓凌峰,李新海,肖立新,等.新型聚合物自由基锂二次电池正极材料[J].电池,2004,34(2):93-95.
[25]刘承梅,陈剑,衣宝廉.有机自由基聚合物PTMA电极的研究与制备[J].电源技术,2012,36(4):458-462.
[26]Shirakawa H,Louis E J,MacDiarmid A G,et al.Synthesis of electrically conducting organic polymers:Halogen derivatives of polyacetylene [J].J.Chem.Soc.Chem.Commun.,1977:578-580.
[27]Nigrey P J,MacDiarmid A G,Heeger A J.Electrochemistry of polyacetylene,(CH)x:Electrochemical doping of (CH)xfilms to the metallic state [J].J.Chem.Soc.Chem.Commun.,1979:594-595.
[28]钱人元,曹镛.有机晶体中的电子过程[M].上海:上海科学技术出版社,1987:261.
[29]Shirakawa H.Handbook of Conducting Polymers[M].2 eds.New York:Marcel Dekker,Inc.,1998:197-208.
[30]Chaing J C,MacDiarmid A G.‘Polyaniline’:Protonic acid doping of the emeraldine form to the metallic regime [J].Synthetic Metals,1986,13:193-205.
[31]Chen Show-An,Lin Liang-Chang.Method for preparing and using electroconductive polymer composites as positive electrode active materials to prepare secondary batteries:US, 5849045[P].1998-12-15.
[32]Chen Show-An,Lin Liang-Chang.Electroconductive polymer composites for use in secondary batteries as positive electrode active materials:US,5863454[P].1999-01-26.
[33]Genies E,Hany P,Santier Ch.Secondary organic batteries made with thick free standing films of electrochemically prepared polyaniline[J].Synth.Met.,1989,28:647-652.
[34]Chu MayYing.Rechargeable positive electrodes:US,5814420[P].1998-09-29.
[35]Liu M,Visco S J,De jonghe L C,et al.Electrochemical properties of organic disulfide/thiolate redox couples[J].Electrochem.Soc.,1989,136(9):2570-2575.
[36]Naoi K,Menda M Ooike H.An enhanced redox process of disulfide compounds at polyaniline film electrode[J].Journalof Electroanalytical Chemistry and Interfacial Electrochemistry,1991,318(2):395-398.
[37]Tatsuma T,Matsui H,Shouji E,et al.Reversible electron transfer reaction bewteen polyaniline and thio/disulfide couples[J].Phys.Chem.,1996,100(33):14016-14021.
[38]Naoi Katsuhiko,Kawase Kenchi,Mori Mitsuhiro,et al.Electrochemistry of Poly (2,2’-dithiodianiline):A new class of high energy conducting polymer interconnected with S—S bonds[J].Electrochem.Soc.,1997,144(6):L173-L175.
[39]Uemachi H,Iwasa Y,Mitani T.Poly (1,4-phenylene-1,2,4-3',5'-yl):The new redox system for lithium secondary batteries[J].Electrochimica Acta,2001,46(15):2305-2312.
[40]Gorkovenko A,Skotheim T A.Electroactive,energy-storing,highly crosslinked,polysulfide-containing organic polymers for use in electrochemical cells:WO,99/33130[P].2001-03-13.
[41]Gorkovenko A,Skotheim T A.Electroactive,energy-storing,highly crosslinked,polysulfide-containing organic polymers and methods for making same:US,6201100[P].2001-03-13.
[42]杨裕生,王维坤,苑克国,等.锂电池正极材料有机多硫化物的展望[J].电池,2002,32(s1):1-8.
[43]李晓林,李琰,王维坤,等.新型锂电池正极材料多硫化碳炔的研究[J].北京化工大学学报,2007,34(4):401-404.
[44]Zhang Shichao,Zhang Lan,Yu Jinhua.Preparation and electrochemical properties of polysulfide polypyrrole[J].Journal of Power Sources,2011,196:10263-10266.
猜你喜欢
新能源汽车供能技术(2021年1期)2021-10-14 08:59:48
电子制作(2019年23期)2019-02-23 13:21:36
广州大学学报(自然科学版)(2015年4期)2015-12-23 11:50:10
大连工业大学学报(2015年4期)2015-12-11 04:06:50
智能建筑电气技术(2015年5期)2015-12-10 05:52:23
中国塑料(2015年7期)2015-10-14 01:02:34
电源技术(2015年5期)2015-08-22 11:18:02
发明与创新(2015年30期)2015-02-27 10:39:51
中国塑料(2014年11期)2014-10-17 03:07:18
技术与教育(2014年2期)2014-04-18 09:21:33
