
基于吩噻嗪类化合物的新型抗结核小分子化合物的设计、合成及活性研究
2013-07-13 01:24季兴跃吴林韬李思阳易红金洁李卓荣
中国医药生物技术 2013年2期
季兴跃,吴林韬,李思阳,易红,金洁,李卓荣
基于吩噻嗪类化合物的新型抗结核小分子化合物的设计、合成及活性研究
季兴跃,吴林韬,李思阳,易红,金洁,李卓荣
100050 北京,中国医学科学院北京协和医学院医药生物技术研究所有机化学室
借助计算机虚拟筛选技术设计并合成新型抗结核小分子化合物。
以氯丙嗪为模板分子,利用 Discovery studio 3.0 构建相应的 3D 药效团模型,并对虚拟化合物库进行筛选;对筛选结果进行手动择优选择,化学合成目标化合物并进行体外抗结核分枝杆菌的活性评价。
筛选得到活性化合物 15 个,并合成其中 13 个,其中化合物 9(多巴胺)在体外对结核分枝杆菌表现出中等强度的抑制作用,MIC 为 8.0 μg/ml。
通过药效团的方法筛选得到了结构较吩噻嗪类化合物简单的抗结核活性小分子多巴胺,该化合物作为抗结核分枝杆菌的先导化合物,较吩噻嗪类具有更大的结构修饰空间。
抗结核药; 计算机辅助设计; 多巴胺; 吩噻嗪
由结核分枝杆菌引起的结核病至今仍然是一个全球性的健康问题。据估计,全球有近 1/3 的人口感染结核分枝杆菌,并且每年大约有 800 万新增感染人群,约 200 万人死于结核病[1-2]。虽然目前的治疗方案几乎能治愈所有药敏性结核病,但仍存在很大缺陷,如治疗周期长(6 ~ 9 个月)以及严重的药物毒副作用。较长的治疗周期严重降低了患者的依从性,从而可能导致结核杆菌耐药,甚至是多药耐药(MDR)。而一旦出现耐药,一线药物,如异烟肼、利福平、乙醇丁胺和吡嗪酰胺[3],就不再有效,只能求助于二线药物,如氟喹诺酮、卷曲霉素、卡拉霉素等。但是这些二线药物不仅疗效差,而且都存在严重的毒副作用[4]。近年来,随着广泛耐药(XDR)结核病菌的出现[5-6],以及结核病与艾滋病的双重感染使得研发具有全新结构类型的抗结核药物愈来愈迫切。
吩噻嗪类化合物(图 1)是一类用于治疗精神病的药物,文献报道其在体内外均具有明显的抑制结核病菌的活性,最小抑菌浓度(MIC)在 0.9 ~ 32 μg/ml[7]。并且,该类化合物对多药耐药以及广泛耐药的结核病菌也表现出一定的抑制活性,且能增强一些一线药物的治疗效果[7]。然而,由于该类化合物抗结核病菌的活性不够强,而且长期服用会导致毒副作用,使得该类化合物在临床上用于治疗结核病受到严重的限制。为了克服这些缺点,很有必要对其进行结构优化以提高其抗结核活性及降低毒副作用。但是,这类化合物普遍具有较高的亲脂性,即便是通过优化得到体外活性较好的化合物,很可能因为其水溶性差以及其他一些不理想的药代动力学属性而很难继续研究开发。所以,较好的选择是以该类化合物的三维结构作为参考,设计筛选出结构简单的苗头(hits)或者先导(lead)化合物,再进行进一步的结构优化。
本论文即是基于吩噻嗪类化合物的抗结核活性及其结构特征,应用虚拟筛选技术(virtual screening)来寻找结构简单的具有抗结核活性的新型先导结构。
1 材料与方法
1.1 材料
1.1.1 分子模拟软件 药效团的构建以及化合物库的虚拟筛选均通过美国Accelrys 公司的 Discovery studio 3.0(DS 3.0)软件中的相关模块来完成。
1.1.2 试剂及仪器 所有试剂(包括目标化合物9 和 10)均为市售化学纯或分析纯,购于北京百灵威公司;MP90 型自动熔点仪为瑞士 Mettler Toledo 公司产品,温度未校正;MERCURY-400 型核磁共振仪为美国 Varian 公司产品,内标为 TMS;AutoSpec Ultima-Tof 串联质谱仪为英国 Micromass 公司产品。

图 1 吩噻嗪类化合物的化学结构
Figure 1 The structure of several known phenothiazines
1.2 方法
1.2.1 药效团的构建及虚拟筛选策略 由于吩噻嗪类化合物抗结核作用的靶蛋白未知,所以不能应用对接(docking)的方法来虚拟筛选。因此,选用基于配体(药效团)的技术来进行虚拟筛选。由于与吩噻嗪类化合物具有一致的抗结核作用机制,并且结构类型完全不同的化合物不可得,所以在构建 3D 药效团的时候,我们选用氯丙嗪(cholorpromazine)作为模板分子,并假设其最低能量构象为活性构象,应用 DS 3.0 软件包中的“Auto pharmacophore generation”模块来自动生成药效团。具体方法如下:氯丙嗪的化学结构由ChemDraw 构建并另存为mol 格式,导入DS 3.0 中,利用“Full minimization”模块对其进行能量最小化,Max steps 设为1000,其他参数设定为缺省值,得到最低能量构象。再利用“Auto pharmacophore generation”产生3D 药效团。并对其进行适当的修改以用于化合物库的虚拟筛选。我们用该药效团作为提问式(query)对本室虚拟化合物库进行筛选。先应用DS 3.0 中的“Ligand pharmacophore mapping”进行虚拟筛选,“Conformation generation”设定为“fast”,“Maximum omitted features”设定为“1”,其他参数设定为缺省值。根据Linpinski 和 Veber 原则去掉筛选结果中成药性差的化合物。同时,为了降低吩噻嗪类化合物神经系统方面的副作用,对得到的化合物进行了药代动力学属性的评价(ADMET),过滤掉易透过血-脑脊液屏障和具有潜在肝毒性的化合物,最终我们得到大约 300 个化合物。随后我们根据匹配值(Fitvalue)、化合物的多样性以及可合成性对这 300 个化合物进行手动的择优挑选。
1.2.2 目标化合物的合成 通过虚拟筛选得到目标化合物 15 个,其中化合物 9 和 10 通过购买得到,其他 13 个化合物均为本实验室合成,合成路线如图 2 ~ 5。
化合物 1a ~ 1c 经过相应的酰氯与各种脂肪胺在二氯甲烷中,以三乙胺为缚酸剂缩合得到,其中化合物 1b由相应的 N-叔丁氧基羰基衍生物 1b' 经三氟乙酸脱保护得到(图 2)。

A:CH2Cl2、Et3N 和各种取代胺,0 ℃~ 室温,收率70% ~ 80%;B:CH2Cl2、CF3COOH,室温,收率 85%
Figure 2 Synthesis of compounds 1a - 1c
化合物 2a ~ 2f 由相应的取代芳胺与氯乙酰氯缩合反应得到,其在碱性条件下,与吗啉发生亲核取代反应得到目标产物 3a ~ 3f(图 3)。
化合物 4' 与二氯亚砜加热回流,并以 N,N-二甲基甲酰胺为催化剂得到中间体化合物 4,化合物 4 再与各种脂肪胺发生亲核取代反应得到目标产物 5a ~ 5b(图 4)。

A:CH2Cl2、Et3N 和氯乙酰氯,0 ℃~室温,收率 70% ~80%;B:无水 K2CO3、DMF,60 ℃,收率 70% ~80%
Figure 3 Synthesis of compounds 3a - 3f

A:SOCl2和 DMF,回流,2 h,收率 90%;B:K2CO3和 DMF,60 ℃,3 h,收率 70% ~ 80%
Figure 4 Synthesis of compounds 5a and 5b
邻氨基苯甲酸甲酯与苯甲酰氯缩合得到化合物 6。为了实现化合物 6 酯基的选择性水解,采用氢氧化钠为碱,四氢呋喃和水做混合溶剂,在室温反应得到了选择性水解产物化合物 7。以 DIC为缩合剂,化合物 7 再与脂肪胺缩合得到目标产物 8a 和 8b(图 5)。

A:苯甲酰氯、CH2Cl2、Et3N,0 ℃~室温,收率 80%;B:NaOH、THF/H2O,室温,收率 85%;C:DIC、CH2Cl2,室温,收率 75%
Figure 5 Synthesis of compounds 8a and 8b
1.2.3 抗结核分支杆菌活性实验 Alamar blue 法检测化合物抑制结核分枝杆菌标准株H37Rv 复制活性。异烟肼和利福平作为阳性对照。受测化合物储存液浓度为64 μg/ml,最终稀释浓度为 0.5 ~ 32.0 μg/ml。用 7H9 培养基进行 2 倍稀释后接种于 96 孔板中,每孔 100 μl。每孔加入 100 μl 菌悬液,使其最终滴度为 1 × 106CFU/ml。将 96 孔板置于 37 ℃条件下培养 5 d,每孔加入 Alamar blue 溶液后,酶标仪测定各孔荧光值。MIC 定义为使荧光值用溶剂对照组校正后相对生长下降≥ 90% 的最低药物浓度。
2 结果
2.1 药效团的构建
以氯丙嗪为模板分子,通过 DS 3.0 中的“Auto pharmacophore generation”模块产生 3D 药效团,得到了如图 6a 所示的 3D 药效团模型,其包含2 个疏水中心(蓝色)、一个氢键受体(绿色)、一个芳香环中心(橙色)以及一个正电荷中心(红色)。
考虑到在吩噻嗪环的 2 位上没有取代基的其他化合物也具有体外抗结核分枝杆菌的活性,如甲地嗪和甲硫哒嗪(图 1),因此我们将药效团6a中与氯原子相对应的疏水中心删掉,得到6b 所示的药效团,并以此作为提问式(query)对化合物库进行虚拟筛选。
2.2 目标化合物的合成
对 13 个目标化合物进行了合成制备,结构均经过1H-NMR 谱和高分辨质谱的确证。所有目标化合物的编号及化学名如表 1 所示。

6a:以氯丙嗪为模板分子在 DS 3.0中构建得到的 3D 药效团;6b:删除 6a 中一个疏水中心特征得到的 3D 药效团
Figure 6 The 3D pharmacophore model
2.3 体外抗结核分枝杆菌活性
Alamar blue 法检测化合物抑制结核分枝杆菌标准株 H37Rv 复制活性。其最小抑菌浓度(MIC)结果如表 2 所示。
表 1 目标化合物编号及化学名
Table 1 The serial number and chemical name of the target compounds

化合物编号Compound No.化学名Chemical name 1aN-(2-吗啉乙基)-3-苯基-5-甲基异噁唑-4-甲酸胺 1b3-(3'-苯基-5'-甲基异噁唑-4'-甲酰胺基)丙胺 1cN-[3-(二甲胺基)丙基]-3-苯基-5-甲基异噁唑-4-甲酰胺 3a2-(2'-吗啉乙酰胺基)-4,5,6,7-四氢苯并[b]噻吩-3-甲酸甲酯 3b2-(2'-吗啉乙酰胺基)苯甲酸甲酯 3c3-(2'-吗啉乙酰胺基)苯甲酸甲酯 3d2-(2'-吗啉乙酰胺基)噻唑-4-甲酸乙酯 3e2-(2'-吗啉乙酰胺基)噻吩-3-甲酸乙酯 3f5-(2'-吗啉乙酰胺基)异酞酸二甲酯 5a4-[3-(二甲氨基)丙氨基]-5-甲基噻唑酮[2,3,-d]嘧啶-6-甲酸乙酯 5b4-(3-吗啉丙氨基)-5-甲基噻唑酮[2,3,-d]嘧啶-6-甲酸乙酯 8aN-[3-(二甲胺基)丙基]-2-苯甲酰氨基苯甲酰胺 8bN-(3-吗啉丙基)-2-苯甲酰氨基苯甲酰胺
由表 2 可看出,虽然所有化合物与药效团匹配的 Fitvalue 值都较高,但是大部分评价的化合物在体外对所测菌株并未表现出明显的抑制活性,仅化合物 9 表现出了中等的抑菌活性(MIC =8.0 μg/ml)。如图7 所示,其与氯丙嗪能较好的叠合。与化合物 9 结构相似的化合物 10 也未表现出明显的抑菌活性,对比两者与药效团匹配的构象可看出(图 8),两者在与药效团特征元素的匹配方面并没有太大的差异,区别在于非药效团部分的取代基不一致:化合物 10 苯环上少了一个羟基取代;苄位多了一个羟基取代以及氨基多了一个甲基取代。由于化合物 9 中的羟基并不是药效团特征,所以导致化合物10 活性消失的原因可能是由于苄位的羟基取代或者氮上的甲基取代,但是由于大多吩噻嗪类化合物在侧链氮原子上也有烷基取代,因此可推测,化合物 10 活性消失可能和其苄位多了一个羟基取代有关。关于该化合物的构效关系有待进一步研究确证。
表 2 目标化合物在体外抑制 MTB H37Rv ATCC 27294 菌株活性
Table 2 The MIC values of target compounds against MTB H37Rv ATCC 27294
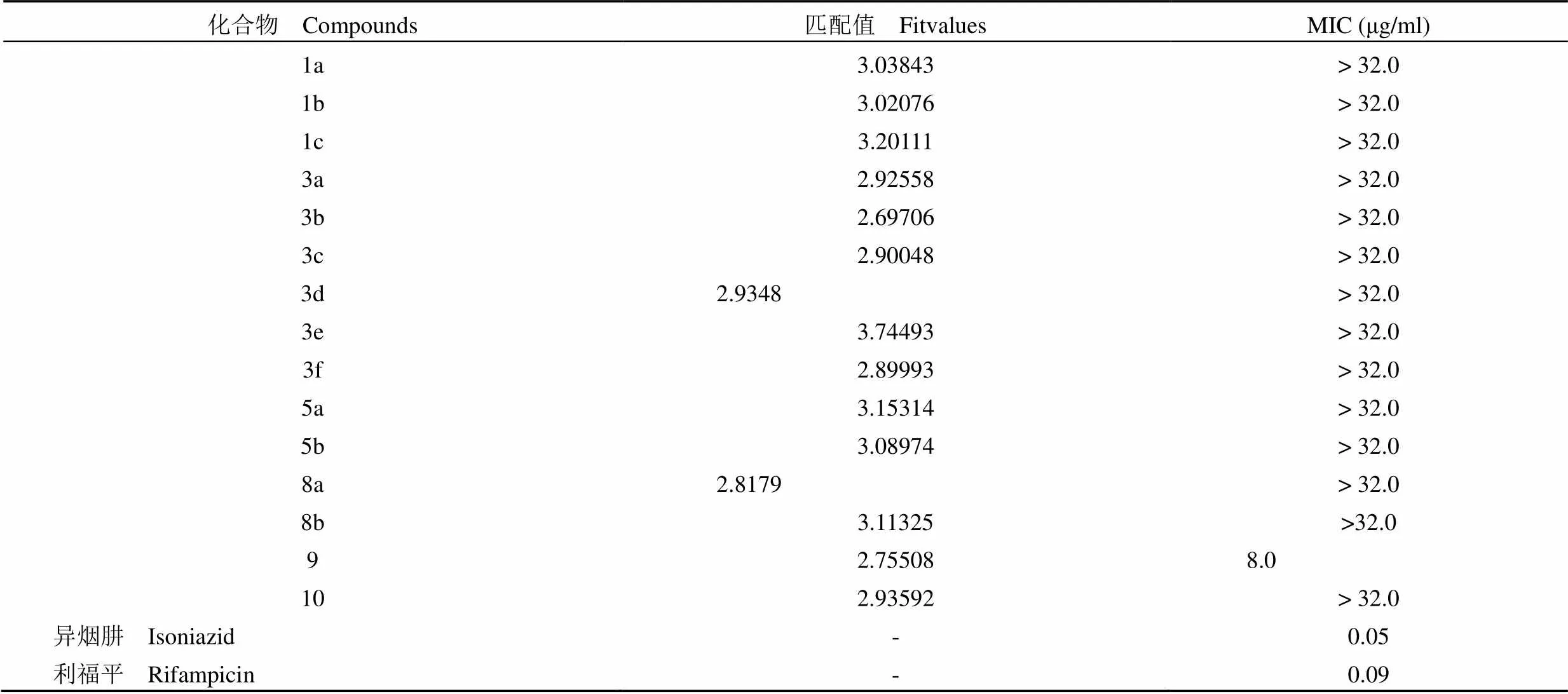
化合物 Compounds匹配值 FitvaluesMIC (μg/ml) 1a3.03843> 32.0 1b3.02076> 32.0 1c3.20111> 32.0 3a2.92558> 32.0 3b2.69706> 32.0 3c2.90048> 32.0 3d2.9348> 32.0 3e3.74493> 32.0 3f2.89993> 32.0 5a3.15314> 32.0 5b3.08974> 32.0 8a2.8179> 32.0 8b3.11325>32.0 92.755088.0 102.93592> 32.0 异烟肼 Isoniazid-0.05 利福平 Rifampicin-0.09
Figure 7 The superimposing conformations of cholorpomazine and compound 9 (green)

图 8 化合物 9 和 10 与药效团匹配的构象
Figure 8 The conformations of compounds 9 and 10 generated by pharmacophore mapping
3 讨论
化合物 9(多巴胺)在体外表现出了一定的抗结核活性。由于多巴胺与吩噻嗪类化合物相比具有较低的分子量和 AlogP 值(分别为 153.178 和 0.773,由 DS3.0 计算得到),且均满足“似先导物三原则”[8-9]所限定的条件(分子量≤ 300;AlogP ≤ 3;不超过 3 个氢键给体及受体)。而对于吩噻嗪类化合物,以氯丙嗪为例,其分子量和 AlogP 值分别为 318.096 和 4.739,均超出了“似先导物三原则”的范围。由于先导物结构优化的过程往往伴随着分子量和 AlogP 的增加[10],而具有高分子量和 AlogP 值的化合物往往具有较差的药代动力学属性,如口服吸收弱,极易被代谢排出体外等。所以吩噻嗪类化合物结构优化的空间相对多巴胺较小,也就是说,多巴胺较之吩噻嗪类化合物“更像先导化合物”。
仅靠药效团来筛选活性化合物是不够的。由药效团得到的结果往往假阳性率较高[11],因为药效团只考虑配体和模型的相互匹配,而没有考虑靶蛋白的三维结构。很多化合物虽然能很好地与药效团匹配,但是其分子结构中非药效团部分很可能与靶蛋白发生不合理的碰撞,从而在体外表现为非活性分子。所以在虚拟筛选的过程中,为了降低此类假阳性率,往往在药效团的基础上增加一个排除体积,或是将药效团初筛得到的结果再进行与靶蛋白的对接,最终以对接结果来作为挑选苗头化合物的标准[12]。但是,增加排除体积和对接都有一个前提,就是活性小分子的作用靶点及其三维结构已知。在本论文中,吩噻嗪类化合物抗结核的作用靶点未知,所以只能用药效团的方法来进行虚拟筛选。另外,由于在构建药效团时,除了吩噻嗪类化合物外,并没有其他结构类型的化合物可用来与吩噻嗪类化合物进行叠合,所以单纯以氯丙嗪作为模板分子产生的药效团难以全面地反映该类化合物抗结核的作用特点。此外,本论文是假设氯丙嗪的最低能量构象为其活性构象,而实际情况可能与此构象存在一定的偏差,所以由此产生的药效团特征在空间上的相对位置可能存在偏差。这些都是本论文筛选得到的化合物假阳性较高的原因。
志谢 感谢北京结核病研究所路宇老师课题组和南京市胸科医院对目标化合物体外抗结核活性的测定。
[1] Bloom BR, Murray CJ. Tuberculosis: commentary on a reemergent killer. Science, 1992, 257(5073):1055-1064.
[2] Wright A, Zignol M, Van Deun A, et al. Epidemiology of antituberculosis drug resistance 2002-07: an updated analysis of the global project on anti-tuberculosis drug resistance surveillance. Lancet, 2009, 373(9678):1861-1873.
[3] Shi R, Itagaki N, Sugawara I. Overview of anti-tuberculosis (TB) drugs and their resistance mechanisms. Mini Rev Med Chem, 2007, 7(11):1177-1185.
[4] O’brien RJ, Nunn PP. The need for new drugs against tuberculosis. Obstacles, opportunities, and next steps. Am J Respir Crit Care Med, 2001, 163(5):1055-1058.
[5] Tamaru A, Nakajima C, Wada T, et al. Dominant incidence of multidrug and extensively drug-resistant specific mycobacterium tuberculosis clones in Osaka prefecture, Japan. PLoS One, 2012, 7(8):e42505.
[6] Li X, Wang H, Jing H, et al. Population-based surveillance of extensively drug-resistant tuberculosis in Shandong Province, China. Int J Tuberc Lung Dis, 2012, 16(5):612-614.
[7] Amaral L, Kristiansen JE, Viveiros M, et al. Activity of phenothiazines against antibiotic-resistant Mycobacterium tuberculosis: a review supporting further studies that may elucidate the potential use of thioridazine as anti-tuberculosis therapy. J Antimicrob Chemother, 2001, 47(5):505-511.
[8] Congreve M, Carr R, Murray C, et al. A ‘rule of three’ for fragment-based lead discovery? Drug Discov Today, 2003, 8(19):876- 877.
[9] Lipinski CA. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today: Technologies, 2004, 1(4):337-341.
[10] Hann MM, Keserü GM. Finding the sweet spot: the role of nature and nurture in medicinal chemistry. Nat Rev Drug Discov, 2012, 11(5): 355-365.
[11] Yang SY. Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov Today, 2010, 15(11-12): 444-450.
[12] Ren JX, Li LL, Zheng RL, et al. Discovery of novel Pim-1 kinase inhibitors by a hierarchical multistage virtual screening approach based on svm model, pharmacophore, and molecular docking. J Chem Inf Model, 2011, 51(6):1364-1375.
Design, synthesis and bioactivity of novelantituberculoticagents based on phenothiazines
JI Xing-yue, WU Lin-tao, LI Si-yang, YI Hong, JIN Jie, LI Zhuo-rong
Department of Medicinal Chemistry, Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100050, China
To design and synthesize novel antituberculotic agents by employing computer aided drug design.
Cholorpromazine was used as modulate compound to generate 3D pharmacophore hypotheses in Discovery studio 3.0, and our in-house compounds library was screened. The candidate compounds were cherry-picked manually for synthesis and evaluation for their anti-TB activity(MTB H37Rv ATCC 27294).
15 active compounds were obtained from the screening and 13 of them were synthesized. The bioassay results showed that compound 9 (dopamine) presented moderately potent inhibitory activity againstwith MIC value of 8.0 μg/ml.
Dopamine was active againstwith lower molecular weight and LogP value. As a lead antituberculotic compound, it shows potential for optimization to improve anti-TB activity by comparison with the phenothiazines.
Antitubercular agents; Computer-aided design; Dopamine; Phenothiazines
10.3969/cmba.j.issn.1673-713X.2013.02.005
“十二五重大新药创制”国家科技重大专项(2012ZX0930 1002-001-017)
李卓荣,Email:l-z-r@263.net
2012-12-07
LI Zhuo-rong, Email: l-z-r@263.net
猜你喜欢
质谱学报(2022年3期)2022-06-15
广州化工(2022年3期)2022-02-24
世界最新医学信息文摘(2021年12期)2021-06-09
中华养生保健(2020年10期)2021-01-18
世界农药(2020年12期)2021-01-04
世界最新医学信息文摘(2020年17期)2020-12-25
中华养生保健(2020年5期)2020-11-16
中华养生保健(2020年7期)2020-11-16
农药科学与管理(2019年8期)2019-11-23
农药科学与管理(2019年12期)2019-05-20
