
Epigenetic origins of metabolic disease:The impact of the maternal condition to the offspring epigenome and later health consequences
2013-05-25 03:20:08RhinnLkerMryWlodekJessiConnellyZhenYn
食品科学与人类健康(英文) 2013年1期
Rhinn C.Lker Mry E.Wlodek Jessi J.Connelly Zhen Yn
a Department of Medicine,University of Virginia,Charlottesville,VA 22908,USA
b Robert M.Berne Cardiovascular Research Center,University of Virginia,Charlottesville,VA 22908,USA
c Department of Physiology,The University of Melbourne,Royal Parade,Parkville,VIC 3010,Australia
Abstract It has long been established that an adverse maternal condition impacts on the developing fetus and predisposes the offspring to develop metabolic and cardiovascular disease in later life.However,the underlying mechanisms that are initiated during development and contribute to the disease predisposition are understudied.Recently,epigenetic reprogramming in early life has emerged as a promising candidate that could cause altered DNA transcription and gene expression into adulthood and contribute to disease susceptibility.This review will focus on the impact of maternal high fat diet to the offspring in early life and the adult health consequences.We will then discuss the current literature supporting a role for epigenetic modification such as DNA methylation and histone modifications as a key mechanism underlying developmental programming.
Keywords:Epigenetics;Developmental programming;Metabolic disease;Maternal high-fat diet;DNA methylation;Histone modification
1.The developmental origins hypothesis
The“developmental origins of adult disease”hypothesis originated from epidemiological studies conducted by David Barker and colleagues in Hertfordshire,England in 1989.They found that being born small for gestational age was a major risk factor for the development of cardiovascular disease later in life with significantl increased the risk of mortality from heart failure[1,2].These studies were the firs of many epidemiological and animal studies that have rigorously investigated the impact of altered fetal,early postnatal and childhood growth on the development of later life diseases.Indeed,it is now unequivocally accepted that early life insults increase the susceptibility of the offspring to developing a plethora of adverse conditions in later life including,but not limited to,type 2 diabetes[3-5],insulin resistance[4-8],hypertension[1]and osteoporosis [9,10].These early life insults to the offspring primarily originate from an adverse maternal condition prior to and/or during pregnancy,such as gestational diabetes,obesity,excessive weight gain,pre-eclampsia,smoking,undernutrition and uteroplacental insufficien y.Each of these abnormal conditions imposes negative impact on the gestational milieu and leads to important consequences for the growth and development of the fetus and health of the offspring in adulthood.
Early life origins of later life disease is often referred to as‘programming’.Define by Hales and Barker,‘programming’is the“permanent or long term change in the structure or function of an organism resulting from a stimulus or insult acting at a critical period of early life”[11].Furthermore,the adult outcomes of early life ‘programming’ are characterized by the nature of the insult and developmental responses,the timing in which the insult occurs,and the duration of the insult[12,13].The insult most adversely affects the organs that undergo rapid growth in the offspring at the time of the insult occurring.Another phenomenon that likely contributes to early life programming events is the“predictive adaptive response”.This occurs when the fetus undergoes or initiates adaptations during gestation or early postnatal development based on the predicted postnatal nutritional environment[14-16].For example,poor nutritional conditions during gestation may trigger metabolic adaptations in the fetus in preparation for poor nutrition after birth in order to ensure survival.However,if the resultant postnatal nutrition is abundant,then the programmed metabolic adaptations that occurredin uterowill become detrimental and likely result in excess energy storage,altered postnatal growth,altered body composition(e.g.increased fat mass:lean mass)and subsequent metabolic dysfunction in later life[11,12,17].
The remainder of this review will primarily focus on 2 important and relevant questions:(1) the impact of maternal obesity and/or high fat diet during gestation on the development of metabolic disease in the adult offspring;and(2)the role of epigenetic modification as the underlying mechanism responsible for the developmental programming of later disease.Although developmental programming is a well-established phenomenon,much of the literature is concerned with the impact of maternal undernutrition and uteroplacental insufficien y that often result in small birth weight and later life disease.However,it is becoming more apparent that the correlation between birth weight and later disease is presented with a U shaped curve;and being born large for gestational age is also a significan risk factor for later disease[18-20].Furthermore,maternal obesity and highfat diet are increasing in prevalence and the impact on offspring health warrants attention.Epigenetics is a relatively new fiel of research that is primarily concerned with the regulation of gene transcription through modification of DNA structure,such as DNA methylation and histone methylation/acetylation.Changes in epigenetic regulation have been shown to occur in early life development in response to environmental cues and unveils new possibilities in the programming fiel as to the long-term impact and mechanistic insight into developmental programming.
2.Maternal obesity/high fat feeding
The prevalence of maternal obesity is increasing at an alarming rate [21],however,the consequences for the health of the offspring in later life is poorly understood compared with that of fetal undernutrition and fetal growth restriction.In various experimental models including sheep and rodents,offspring of obese mothers,or mothers exposed to a high-fat diet generally exhibit increased birth weight and fat mass[22-28].Other reports showed no changes in birth weight[29-37]possibly due to differences of the animal models and experimental designs.Furthermore the severity,length and timing of maternal highfat feeding and the postnatal growth profil of the offspring may also contribute to the interpretation of offspring health outcomes.In later life,offspring of obese or high-fat fed mothers are more susceptible to developing non-alcoholic fatty liver disease [30,38],insulin resistance [39,40],glucose intolerance[24,25,39,40],obesity [25,39,40],hyperphagia [25],hypertension [25,32,41]and cardiovascular impairments [25,32,41].Tissue-specifi alterations in gene expression and organ development in early life,induced by obesity related maternal factors that alter the gestational milieu,likely have impact on tissue function throughout the life span.
Fetal nutrient supply during gestation is dependent on both maternal nutritional status and the transfer of those nutrients across the placenta.Obesity and high-fat diet are often associated with elevated circulating lipids,inflammatio and insulin resistance.During pregnancy,obesity and high-fat diet result in significantl increased lipid transfer across the placenta and exposure of the fetus to lipids at an earlier stage of gestation than normal[42].The early and increased lipid exposure is likely augmented by elevated gene and protein expression of the placental fatty acid transporters (FATP1,FATP4 and CD36),which has been reported in obese pregnant ewes at 75 and 135 d of gestation(term=145 d)[42].Indeed,elevated free fatty acids,triglycerides and cholesterol were also observed in the placenta and in the plasma of the fetus [28].Lipid species are able to activate cell signaling pathways and act as ligands for nuclear receptors.Therefore,increased circulating lipids in the fetal circulation has the potential to alter gene expression during development and may play a role in the cellular signaling processes responsible for increased susceptibility of later disease.
Elevated circulating lipids are associated with widespread inflammatio in obese conditions[43].In obese pregnant ewes,elevated lipids in the placenta was associated with activation ofinflammator signaling pathways of c-Jun N-terminal kinase(JNK/c-jun) and nuclear factor kappa-light-chain-enhancer of activated B cells(NF-κB)viaactivation of the Toll-like receptor 4(TLR4)of which free fatty acids are known ligands[28].Activation ofinflammator signaling was also associated with increased levels of pro-inflammator cytokines including tumor necrosis factor-α (TNF-α),interleukin (IL)-1,IL-6,IL-8 and IL-18 mRNA in the placenta[28].Importantly,similar finding were reported in obese human mothers along with macrophage accumulation in the placenta [23].It is likely that the inflam matory factors and resulting oxidative stress cues within the placenta of obese mothers can be transferred to the fetus and activate/repress important signaling events resulting in abnormal development [23,28].However,to date few studies have investigated the impact of maternal or placental inflammatio on offspring development and later health.One study showed that inflammatio during pregnancy had a significan adverse impact on the development of the fetal brain and nervous system [44],which could contribute to behavioral and cognitive irregularities and conditions such as autism,schizophrenia,cerebral palsy,blindness and mental retardation.It is,therefore,likely that a maternal obesity-induced inflammator environment would contribute to maladaptive programming events in other fetal organs/tissues.Indeed,inflammatio has been shown to impair myogenic differentiation and skeletal muscle development [45,46],while promoting adipogenesis possibly through alterations in NF-κB signaling [26,47].Therefore,maternal-fetal inflammatio represents an important mechanism likely responsible for alterations in the developmental gene program and abnormal growth leading to increased disease susceptibility later in life.The impact of maternal obesity/high fat diet to offspring during development and consequences in later life are summarized in Fig.1.
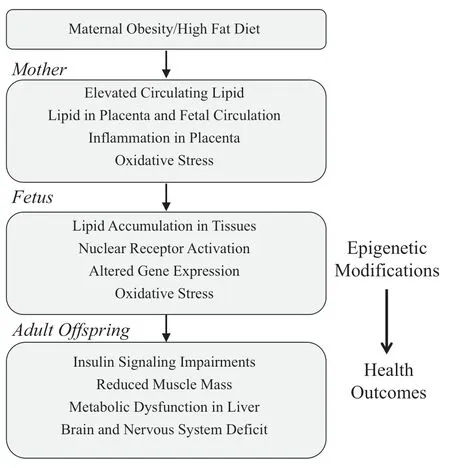
Fig.1.Flow diagram summarizing the characteristics of obesity/high fat diet during pregnancy and the impact on the developing fetus that may contribute to maladaptation and adverse health outcomes in adult offspring.
3.Offspring metabolic outcomes of maternal obesity/high-fat feeding
3.1.Liver
The fetal liver is particularly vulnerable to changes of maternal nutritional status since nutrients that cross the placenta into the fetal circulation are firs shuttled to the liver.In obese pregnancies,the liver is directly exposed to elevated lipid levels with consequential lipid accumulation and alterations of metabolism and gene expression.Buckley et al.[22]reported that maternal high-fat feeding led to elevated liver triglycerides in conjunction with reduced insulin signaling proteins (~50% reduction in insulin receptor β)in rat offspring at 3 months of age along with a subtle impairment of whole body metabolic phenotype[22].Another study demonstrated in rats that long-term maternal high-fat feeding resulted in accumulation of lipid vacuoles in hepatocytes of 36 weeks old offspring and demonstrated a fatty liver phenotype[41].Furthermore,elevations in serum C reactive protein(CRP)were reported in the female offspring of high-fat fed mothers suggesting a role for inflammatio in fetal programming [41].The mechanism by which inflammatio in the mother/fetus causes adverse liver adaptation and later life health consequences is yet to be fully elucidated and warrants further investigation.
3.2.Skeletal muscle
Skeletal muscle plays a major role in whole body insulin sensitivity since it is responsible for about 80%ofinsulin stimulated glucose disposal in the body[48].Skeletal muscle deficit that contribute to whole body metabolic dysfunction include reductions in muscle mass(fat:lean mass ratio)and insulin resistance,both of which have been observed in offspring exposed to an abnormal gestational milieu.In models of fetal undernutrition,impaired skeletal muscle development has been observed in early life [32,49,50]with functional consequences for muscle strength later in life [49,51,52].In the mouse,offspring of obese mothers have reduced muscle mass[25],which is likely a consequence of reduced fibe number[36,47]and/or reduced fibe size[26,36,47].The underlying mechanisms for this abnormal muscle development may be altered transcriptional control of the myogenic regulatory factors(MRFs)that are critical for muscle cell specificatio and differentiation.One study reported reduced expression of myogenic differentiation 1(MyoD)and myogenin,in fetal sheep at day 75 of gestation in mothers fed an obesogenic diet during pregnancy[26].MyoD and myogenin are two MRFs responsible for muscle cell lineage specificatio and differentiation.Therefore,reduced expression of these factors could lead to reduced number of myogenic cells and ultimately result in lower muscle mass in fully developed animals.This may have significan negative impact on metabolic disease in adulthood,especially in the face ofincreased metabolic demand such as occurs with high-fat diet and/or aging.
Programmed impairments in skeletal muscle metabolism,such as changes in the insulin signaling pathway also contribute to the adult metabolic phenotype.For example,Jensen and colleagues showed that adult humans with low birth weight had impaired insulin-stimulated phosphatidylinositol 3-kinase (PI3K)/Akt activation in skeletal muscle [53].Others have shown reduced expression of key proteins in the insulinsignaling pathway in skeletal muscle in adult humans [54,55]and rodents[55]that were born small.Similarly,overnutrition in pregnant ewes [27,56]and high-fat feeding in rodents [57]also resulted in reduced skeletal muscle PI3K activity associated with altered insulin receptor substrate-1 (IRS-1) serine phosphorylation.Another critical regulator of cellular metabolism is the AMP-activated protein kinase(AMPK)which is an energysensing molecule and when activated increases fat oxidation and glucose utilization in the muscle improving insulin sensitivity and glucose uptake [58].Indeed,Zhu and colleagues reported reduced AMPK signaling and impaired development of skeletal muscle in fetuses of obese ewes[27].Taken together,these data suggest that offspring exposed to an abnormal gestational environment are susceptible to reduced skeletal muscle substrate utilization mediated by impairments in multiple signaling pathways.However,not all studies have reported impaired muscle metabolism in offspring exposed to maternal over-nutrition.Buckley and colleagues reported increased expression ofinsulin signaling proteins in the quadriceps muscle as well as normal metabolic phenotype in 3 months old rat offspring born to mothers fed a diet high in polyunsaturated fats [22].Nevertheless,their findin was accompanied by increased total body fat and abdominal fat as well as reduced lean mass[22],which would likely result in metabolic dysfunction later in life.The elevated insulin signaling proteins in the muscle may be compensating for the overall metabolic imbalance,which may not be sustained with age.
The programming of skeletal muscle mass and metabolism in offspring exposed to obese pregnancies is likely related to fetal inflammatio and inflammator signaling in the muscle during development.For example,the reduced expression of MyoD and myogenin in the fetal sheep,as discussed above,were accompanied by increased NF-κB signaling in the skeletal muscle[26].Others have reported elevated skeletal muscle NFκB signaling associated with insulin resistance and increased intramuscular adipocytes in late gestation fetuses of obese ewes[36].Finally,Zhu et al.reported increased TNF-α in the fetal circulation and increased oxidative stress in the muscle of offspring of over-nourished ewes,implicating inflammatio as a potential mediator of developmental programming[27].
In summary,obesity and/or high-fat diet during gestation and early life significantl contributes to metabolic dysfunction in the offspring both in early life and in adulthood when a disease phenotype is often more apparent.Although lipid accumulation and inflammatio in offspring appears to be consistently associated with maternal obesity and high-fat feeding,there has yet to be a definit ve mechanism identifie to confir a causative role.Currently,the leading mechanistic candidate responsible for early life programming of later disease is epigenetic modification induced by a maternal insult that is maintained into adulthood,resulting in altered gene transcription and functional consequences of abnormal metabolic phenotype.
4.Epigenetic mechanisms
Epigenetic modification are structural changes that occur to the DNA without changing the DNA sequence and can result in altered gene expression [59-63].Epigenetic modifi cations of genes have been implicated in the development of many human diseases,including cancers [64-66],neurological disorders [67,68]and type 2 diabetes [69-72].The most well described epigenetic modification are DNA methylation of cytosine residues within cytosine-guanine dinucleotides(CpG islands),and histone modification with methylation and acetylation of the lysine residues in the N-terminal tail of core histones of the chromatin structure.More recently,microRNA regulation of gene expression has also been identifie as a mechanism responsible for epigenetic modification and evidence suggests microRNAs may play a role in regulation of DNA methylation [73,74].These epigenetic marks,particularly if they are near the promoter regions of functional genes,can influenc transcription by altering access of transcriptional machinery to the DNA.DNA methylation may occur through recruitment of Methyl-CpG binding domain proteins (MBDs),while histone modification can change chromatin confirmatio between open and closed states,resulting in altered availability of DNA for transcription.In the past,epigenetic regulation was believed to occur exclusively during germ cell development and in preimplantation embryos and to be sustained throughout the life span[75,76];however,it is now apparent that epigenetic events can occur in response to a variety of environmental cues throughout life and are not necessarily permanent modification [75,76].
Recent evidence supports a role for altered epigenetic regulation to occur in the fetus in response to changes of the gestational milieu[77-82].Abnormal epigenetic regulation may impact on transcriptional control of functionally important genes during development as well as in later life,contributing to abnormal function and disease susceptibility.During periods of tissue/organ development,cells are rapidly dividing and replicating DNA,making the genome more prone to epigenetic regulation.The timing of the maternal insult may have tissuespecifi consequences due to timed tissue development.Stable changes to the epigenome and consequent alterations in gene transcription during early embryogenesis may result in changes to stem cell lineage specification in the rate and number of mitosis and/or apoptosis,in the expression of structural proteins and of the metabolic and homeostatic control processes.Any one or combination of these consequences could cause permanent deficit in organ structure and function and represent a major health burden for the offspring in later life.Table 1 summarizes the current literature reporting on epigenetic modification in different models of developmental programming and their observed or predicted impact on later metabolic health of the offspring.
Another complicating factor in epigenetic research is that DNA methylation and histone modification can interact with and influenc each other[59,83,84].These interactions are not well understood and add an additional layer of complexity in elucidating the mechanism underlying the functional consequences of early programming events.For the purposes of this review,DNA methylation and histone modification will be addressed individually and their interactions will not be considered.The following sections will focus on the existing research in developmental programming of gene and tissue-specifi epigenetic marks that provide insight into the potential underlying mechanisms of later metabolic disease.
4.1.DNA methylation
Methylation of DNA primarily occurs on the 5′position of cytosine bases within CpG dinucleotide pairs,resulting in the formation of 5-methylcytosine.This event is mediated by the DNA methyltransferases(DNMT 1,3a and 3b).DNMT3a and 3b are primarily responsible forde novomethylation events[85],which are maintained following mitosis by DNMT1 [86].The major regulators of DNMT activity are not well defined however,the leading candidates include microRNA regulation[87]and phosphorylation of the DNMT targeting domain[88,89]as well as other post-translational modification [90].Changes in translational activation and protein degradation of DNMTs can also contribute to their abundance and hence total enzymatic activity.Depending on the region of the gene where the methylation occurs,gene transcription can be silenced by the recruitment of Methyl-CpG binding domain proteins(MBDs)to the methylated CpG.The binding of MBDs recruit other factors to the DNA that contribute to alteration of chromatin structure and blocking access of transcription factors to the DNA for initiation of transcription[91].Some of the factors that contribute to the silencing of genes following methylation of CpG sites include the corepressor protein (Sin3a),ATP-dependent remodeling protein(NuRD) and histone deacetylase (HDAC),which all interfere with transcription factors and the binding of RNA Polymerase II to the promoter sequence.Non-CpG methylation(methylation at cytosine bases outside of a CpG site)has also been observed and although less is known about the consequences of non-CpG methylation on gene expression some evidence suggests it may effect transcriptional processes such as RNAsplicing,alternative transcripts and alternative promoters[92-94].
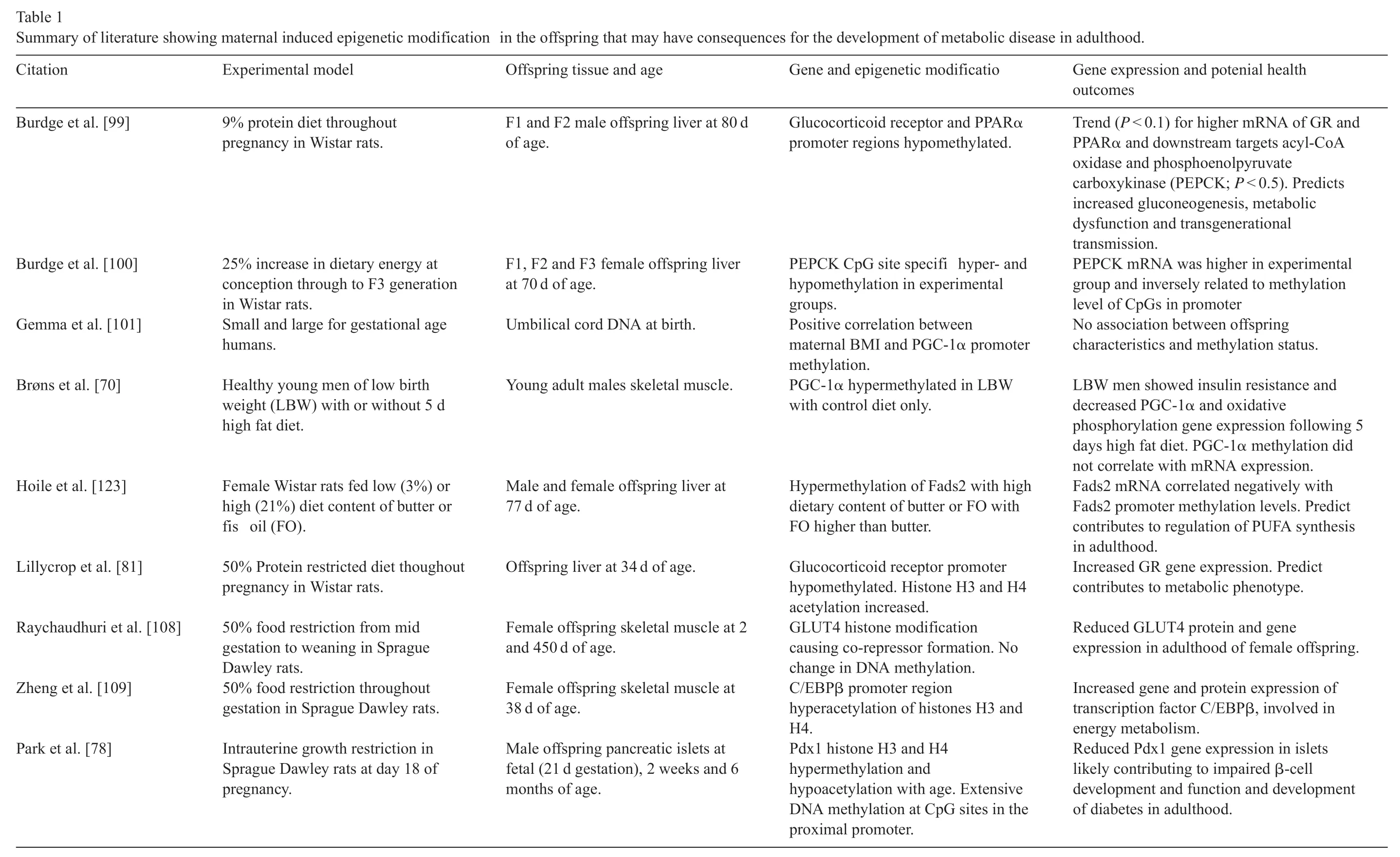
Table 1 Summary of literature showing maternal induced epigenetic modification in the offspring that may have consequences for the development of metabolic disease in adulthood.Citation Experimental model Offspring tissue and age Gene and epigenetic modificatio Gene expression and potenial health outcomes Burdge et al.[99]9%protein diet throughout pregnancy in Wistar rats.F1 and F2 male offspring liver at 80 d of age.Glucocorticoid receptor and PPARα promoter regions hypomethylated.Trend(P <0.1)for higher mRNA of GR and PPARα and downstream targets acyl-CoA oxidase and phosphoenolpyruvate carboxykinase(PEPCK;P <0.5).Predicts increased gluconeogenesis,metabolic dysfunction and transgenerational transmission.Burdge et al.[100]25%increase in dietary energy at conception through to F3 generation in Wistar rats.F1,F2 and F3 female offspring liver at 70 d of age.PEPCK CpG site specifi hyper-and hypomethylation in experimental groups.PEPCK mRNA was higher in experimental group and inversely related to methylation level of CpGs in promoter Gemma et al.[101]Small and large for gestational age humans.Umbilical cord DNA at birth.Positive correlation between maternal BMI and PGC-1α promoter methylation.No association between offspring characteristics and methylation status.Brøns et al.[70]Healthy young men of low birth weight(LBW)with or without 5 d high fat diet.Young adult males skeletal muscle.PGC-1α hypermethylated in LBW with control diet only.LBW men showed insulin resistance and decreased PGC-1α and oxidative phosphorylation gene expression following 5 days high fat diet.PGC-1α methylation did not correlate with mRNA expression.Hoile et al.[123]Female Wistar rats fed low(3%)or high(21%)diet content of butter or fis oil(FO).Male and female offspring liver at 77 d of age.Hypermethylation of Fads2 with high dietary content of butter or FO with FO higher than butter.Fads2 mRNA correlated negatively with Fads2 promoter methylation levels.Predict contributes to regulation of PUFA synthesis in adulthood.Lillycrop et al.[81]50%Protein restricted diet thoughout pregnancy in Wistar rats.Offspring liver at 34 d of age.Glucocorticoid receptor promoter hypomethylated.Histone H3 and H4 acetylation increased.Increased GR gene expression.Predict contributes to metabolic phenotype.Raychaudhuri et al.[108]50%food restriction from mid gestation to weaning in Sprague Dawley rats.Female offspring skeletal muscle at 2 and 450 d of age.GLUT4 histone modification causing co-repressor formation.No change in DNA methylation.Reduced GLUT4 protein and gene expression in adulthood of female offspring.Zheng et al.[109]50%food restriction throughout gestation in Sprague Dawley rats.Female offspring skeletal muscle at 38 d of age.C/EBPβ promoter region hyperacetylation of histones H3 and H4.Increased gene and protein expression of transcription factor C/EBPβ,involved in energy metabolism.Park et al.[78]Intrauterine growth restriction in Sprague Dawley rats at day 18 of pregnancy.Male offspring pancreatic islets at fetal(21 d gestation),2 weeks and 6 months of age.Pdx1 histone H3 and H4 hypermethylation and hypoacetylation with age.Extensive DNA methylation at CpG sites in the proximal promoter.Reduced Pdx1 gene expression in islets likely contributing to impaired β-cell development and function and development of diabetes in adulthood.
4.2.DNA methylation and metabolic disease
Several studies have focused on functionally important metabolic genes to link epigenetic modification with the development ofinsulin resistance or type 2 diabetes.A major candidate identifie that undergoes epigenetic regulation and has been implicated in type 2 diabetes is the master regulator of mitochondrial biogenesis,peroxisome proliferator activated receptor-γ(PPARγ)co-activator-1α(PGC-1α).Epigenetic regulation of PGC-1α has been shown in the skeletal muscle[72]and pancreatic islets of humans with type 2 diabetes [71]and in the liver of patients of non-alcoholic fatty liver disease[95].Barres and colleagues showed in the skeletal muscle of type 2 diabetes patients that methylation of CpG at-260 in the promoter of the PGC-1α gene was hypermethylated,and this hypermethylation correlated with reduced PGC-1α gene expression[72].Importantly,hypermethylation of the PGC-1α promoter was gene-specifi since no differences in DNA methylation were detected on the flankin genes.Interestingly,non-CpG methylation was also found to be elevated in the PGC-1α gene and was acutely increased in human myotubes following treatment with TNF-α or free fatty acids [72]and provides a possible functional link between epigenetic modificatio and disease phenotype.Furthermore,using siRNA knock down of each DNMT isoform(1,3a and 3b)in primary human myocytes,Barres and colleagues identifie DNMT3b as the isoform responsible for palmitate-induced methylation of PGC-1α[72].
Transgenerational transmission of epigenetic marks was firs described by Morgan and colleagues using the Avy-dependent coat color mouse[96].The Avy allele carries an intra-cisternal A particle retrotransposon(IAP)with a cryptic promoter region upstream of the agouti gene.The transcription of the agouti gene originates from the IAP resulting in constitutive expression of the agouti protein,and the mice display a yellow agouti phenotype.Morgan and colleagues showed that methylation of the IAP region was associated with a wild-type coat color phenotype,even though the mice were genetically agouti.In addition,parents that displayed an agouti phenotype were more likely to produced agouti offspring,while parents displaying a wild-type phenotype were more likely to produced offspring of wildtype phenotype.This study suggested that DNA methylation marks could be transmitted across multiple generations [96].Other studies utilizing the Avy mouse showed that methyl supplemented diets during pregnancy were sufficien to increase methylation of the Avy-allele and produce offspring of pseudoagouti appearance regardless of the parents phenotype[97,98],and this phenytope was then passed on to the F2 generation[98].These were the firs studies showing that manipulations of the maternal condition could cause epigenetic modification in the offspring and produce a variety of phenotypic outcomes even though the offspring were genetically identical.
Multiple studies have since shown that other manipulations to the maternal condition can also impact on DNA methylation status of the offspring.For example,maternal dietary protein restriction in pregnant rats has been shown to reduce promoter methylation in the offspring liver of the nuclear receptors peroxisome proliferator activated receptor α(PPARα)and glucocorticoid receptor (GR),of which the target genes play key roles in glucose and lipid metabolism [99].Indeed,these methylation changes were associated with changes in target gene expression and glucose and lipid metabolism[99].Furthermore,hypomethylation of PPARα and GR was also observed in the F2 generation,supporting the premise that epigenetic changes can be inherited and thus increased susceptibility to metabolic disease may also be transmitted across generations through epigenetic mechanisms[99].Importantly,in a separate study in rat offspring of protein restricted dams,hypomethylation of GR in the liver was associated with reduced DNMT1 expression and binding to the GR promoter[81].In a more recent study by the same group,they found that a 25% increase in dietary energy from conception in F0 female rats induced altered methylation of specifi CpG islands in the phosphoenolpyruvate carboxykinase (PEPCK) gene in the liver of the F1 generation along with increased DNMT3a expression[100].PEPCK is the ratelimiting enzyme for gluconeogenesis in the liver,and altered methylation status of PEPCK in the F1 generation was associated with increased mRNA levels of the PEPCK gene,indicative ofincreased liver gluconeogenesis[100].Interestingly,when the 25%increase in dietary energy was maintained throughout the life span of the F2 and F3 generations,some but not all epigenetic changes observed in the F1 generation were observed in successive generations.Therefore,DNA methylation marks may only be maintained into later generations depending on the specifi CpG site,which may be dependent on location within the gene and recruitment of other binding factors.
Since the PGC-1α promoter has been shown to be hypermethylated in skeletal muscle and pancreatic islets of patients with type 2 diabetes,an important question now is whether these marks were established during fetal development?Studies in humans have shown that maternal BMI positively correlated with PGC-1α methylation in umbilical cord DNA [101],while others showed in men that were born small and hence were growth restricted during gestation that PGC-1α was hypermethylated in skeletal muscle [70].However,in this study,hypermethylation of PGC-1α in the muscle had no apparent functional impact,and only following 5 days of high-fat diet feeding was PGC-1α gene expression reduced compared with normal birth weight,age-matched controls on high fat diet[70].These finding suggest that a second insult may be required to unmask the consequences of epigenetic modification on gene expression and phenotypic outcomes.Indeed,the health implications of an abnormal gestational milieu are more apparent when there is additional metabolic demand in postnatal life,such as accelerated growth during childhood,high fat diet or aging[5,102].
4.3.Histone modification
Histone modification include a variety of post-translational modification with the most prominent being acetylation/deacetylation and methylation/de-methylation of the lysine residues in the N-terminal tails.These modification alter gene transcription by changing the confirmatio of the chromatin into open or closed states and hence alter availability of gene promoter regions to transcriptional machinery.Acetylation of histones leads to an open chromatin confirmatio and is regulated by the enzymes histone acetyl transferase(HATs)/histone deacetylase(HDACS)while methylation of histones promotes a closed confirmatio and is regulated by histone methyl transferase(HMTs)/histone demethylase(HDM)[103,104].
In the developmental programming field some early studies identifie histone modification in rat offspring exposed to uteroplacental insufficien y during gestation with overall hyperacetylation of histone H3 in the liver at birth [105].These modification were associated with specifi changes in the expression of metabolic regulatory gene,PGC-1α,and lipid metabolism intermediate transporter gene,carnitine palmitoyltransferase 1 (CPT1) along with site-specifi hyperacetylation of histone H3 lysine K9 in the promoter regions of these genes [105].More recent studies investigating maternal obesity reported association of histone modification in perinatal primate offspring with fatty liver disease [106],while others have showed histone modification in the PEPCK gene in the liver associated with increased gluconeogenesis in neonatal rat offspring of high-fat fed mothers [107].Whether these modification are sustained into later life and ultimately contribute to disease pathogenesis has yet to be determined.Nevertheless,these finding have highlighted the possibility of maternal-induced changes to offspring liver metabolism through modification to the histone code.
Much of the interest in developmentally induced alterations in epigenetic marks and later disease development has been focused on the liver with only a handful of studies investigating histone modification in skeletal muscle.It is known that skeletal muscle metabolic regulation is impaired in growth restricted rats and humans,which is associated with reduced expression of the insulin responsive glucose transporter 4(GLUT4)[53-55].These changes in GLUT4 expression have recently been shown to be mediated through histone deacetylation and di-methylation at sites in the GLUT4 gene causing co-repressor complex formation resulting in decreased GLUT4 transcription at birth that persists into adulthood[108].Interestingly,the histone modification that caused altered GLUT4 transcription were found to be in the absence of any changes to DNA methylation [108].Others showed skeletal muscle-specifi hyperacetylation of histones H3 and H4 in rats exposed to maternal dietary protein restriction in the promoter region of CCAAT/enhancer-binding protein(C/EBPbeta)[109].C/EBPbeta regulates the expression of genes involved in energy homeostasis and muscle development;however,the functional consequences for skeletal muscle metabolism and function were not further investigated.
Tissue-specifi epigenetic modification have also been demonstrated in pancreatic islets in rats exposed to intrauterine growth restriction.The pancreatic and duodenal homeobox 1 (Pdx1) gene is a transcription factor essential for pancreatic β-cell development and function,and its expression was found to be reduced in growth restricted rats at birth as well as in adulthood and was associated with the onset of diabetes [78].The functional outcomes were attributed to altered acetylation and methylation states of the H3 and H4 histones as well as hypermethylation of the CpG island proximal to the promoter of the Pdx1 gene [78].These finding in the skeletal muscle and pancreas support a role for tissue-specifi epigenetic modification in offspring induced by an altered maternal condition and their contribution to tissue-specifi phenotype and disease development.
4.4.MicroRNA
MicroRNAs are small non-coding RNAs that can affect transcriptional processing of genes.Some studies have found that microRNAs may play a role in the regulation of DNA methylation levels [73,74].In one study,Dicer1 deficien mice,a molecule involved in the generation of silencing microRNAs,were found to have reduce expression of a cluster of microRNAs-290 which was associated with defects in DNA methylation and reduced expression of DNMT1,3a and 3b isoforms[73].
Since the role of microRNAs in epigenetic regulation is an emerging concept in a relatively new field few studies in developmental programming have investigated microRNA changes in offspring development.One study performed a microarray in the liver of rat offspring from mothers fed a high-fat diet and reported that 23 microRNAs were reduced between 1.5 and 5-fold including microRNA-709,which is the most abundantly expressed microRNA in the liver [110].Interestingly,methyl CpG binding protein 2(MECP2)and MBD6 are major targets for microRNA-709 and these finding provide a potential link between developmental programming and epigenetic modifica tions mediated through microRNA regulation.
4.5.Potential therapeutic strategies
Some studies have aimed at identifying interventions to prevent adverse health outcomes for offspring exposed to poor developmental conditions.Some have investigated the potential benefit ofimproved nutrition after birth[111,112]while other studies have investigated the impact of exercise in the offspring at different ages[113-119].These studies were performed in models of fetal growth restriction and,to our knowledge,there have been no studies aimed at offsetting the adverse consequences of maternal high fat diet on offspring health.However,these intervention studies provide important information that could be applied to other adverse maternal conditions and the consequences for the offspring.A series of studies conducted in the Wlodek laboratory investigated the potential ofimproving postnatal nutrition by cross-fostering rat offspring that were growth restrictedin uteroonto a healthy mother that produced better quality lactation[111,120].Their major finding showed that improved postnatal lactation in small birth weight offspring partially ameliorated the impaired glucose tolerance[111]and normalized pancreatic β-cell deficit at 6 months of age.Others reported that skeletal muscle mtDNA content at 15 and 20 weeks of age was partially rescued in rat offspring exposed to maternal protein restriction throughout gestation and lactation,when weaned onto a control diet at 4 weeks of age compared with litter mates weaned onto a low protein diet[112].
Recently,the potential benefit of exercise training in the offspring have also generated interest in the developmental programming field Earlier studies in humans showed that regular moderate exercise protected elderly people born of low birth weight from glucose intolerance [118].A more recent study in humans showed that 9 days ofintensive exercise training improved mitochondrial function in adult offspring of mothers with type 2 diabetes [119].However,improvements in insulin sensitivity were only observed in offspring from healthy mothers[119].It is possible that an intervention in early life when the offspring are undergoing rapid growth and development would more likely produce long lasting consequences to later health outcomes.Indeed studies in rats exposed to maternal nutrient restriction[115]during gestation and then exercised in the early juvenile period demonstrated subtle metabolic improvements and increased insulin stimulated GLUT4 translocation to the plasma membrane in adulthood[115].Furthermore,in rat offspring exposed to uteroplacental insufficien y,early exercise training had a remarkable‘reprogramming’effect to normalize pancreatic islet surface area and β-cell mass at 6 months of age[113].Finally,the impact of maternal exercise in mice and rats prior to mating as well as throughout pregnancy and nursing on adult offspring metabolic outcomes was recently reported by Carter and colleagues[121,122].In both mice and rats,mature offspring of dams that exercised displayed improved whole body metabolic phenotype as well as increased glucose disposal in skeletal muscle [121,122],representing a promising intervention that may influenc epigenetic mechanisms during birth that have significan impact on later metabolic health.Importantly,since the role of epigenetic reprogramming is a recent focus in the fiel there have been no studies to determine whether any of the aforementioned interventions were associated with an epigenetic mechanism and represents an important next step in intervention studies.
5.Conclusion
Although currently sparse,there is a growing abundance of literature to support epigenetic modification as underlying mechanisms to developmental programming with long-term functional consequences for disease development in adult life.The current studies are limited in their interpretation,and many of the finding demonstrate associations without confirmin sufficien y or necessity for epigenetic modification in later disease development.Furthermore,epigenetic regulation involves complex protein-DNAinteractions,presenting significan challenges to experiment execution and data interpretation.Nevertheless,epigenetic modification will likely be the key players in the underlying mechanisms of developmental programming and will be fundamental to future research in this field

- 食品科学与人类健康(英文)的其它文章
- Black tea in chemo-prevention of cancer and other human diseases
- Changes in physicochemical properties of proteins in Kayserian Pastirma made from the M.semimembranosus muscle of cows during traditional processing
- Polyphenolic extract of Sorghum bicolor grains enhances reactive oxygen species detoxificatio in N-nitrosodiethylamine-treated rats
- Protective role of concomitant administration of fla lignan concentrate and omega-3-fatty acid on myocardial damage in doxorubicin-induced cardiotoxicity
- GUIDE FOR AUTHORS
- Natural products for cancer prevention associated with Nrf2-ARE pathway