
Identification and functional analysis of miRNAs in developing kernels of a viviparous mutant in maize
2013-05-08 08:24:42HipingDingJinGoMoLuoHuPengHijinLinGungshengYunYouShenMojunZhoGungtngPnZhimingZhng
The Crop Journal 2013年2期
Hiping Ding,Jin Go,Mo Luo,Hu Peng,Hijin Lin,Gungsheng Yun, You Shen,Mojun Zho,Gungtng Pn,*,Zhiming Zhng,*
aInstitute of Maize Research,Key Laboratory of Biology and Genetic Improvement of Maize in Southwest Region,Ministry of Agriculture, Sichuan Agricultural University,Wenjiang 611130,China
bDrug Discovery Research Center of Luzhou Medical College,Luzhou Medical College,Luzhou 3344789,China
cSichuan Tourism University,Chengdu 610041,China
dCollege of Life and Basic Science,Sichuan Agricultural University,Ya'an 625014,China
Identification and functional analysis of miRNAs in developing kernels of a viviparous mutant in maize
Haiping Dinga,1,Jian Gaoa,1,Mao Luob,Hua Pengc,Haijian Lina,Guangsheng Yuana, Yaou Shena,Maojun Zhaod,Guangtang Pana,*,Zhiming Zhanga,*
aInstitute of Maize Research,Key Laboratory of Biology and Genetic Improvement of Maize in Southwest Region,Ministry of Agriculture, Sichuan Agricultural University,Wenjiang 611130,China
bDrug Discovery Research Center of Luzhou Medical College,Luzhou Medical College,Luzhou 3344789,China
cSichuan Tourism University,Chengdu 610041,China
dCollege of Life and Basic Science,Sichuan Agricultural University,Ya'an 625014,China
A R T I C L E I N F O
Article history:
Received 18 April 2013
Revised 3 May 2013
Accepted 17 July 2013
Available online 6 August 2013
Ear germination-associated miRNAs
Given the important roles of miRNAs in post-transcriptional gene regulation,identification of differentially expressed miRNAs will facilitate the elucidation of molecular mechanisms underlying kernel development.In this study,we constructed a small RNA library to comprehensively represent the full complement of individual small RNAs and to characterize miRNA expression profiles in pooled ears of maize(Zea mays L.)at 10,15, 20,22,25 and 30 days after pollination(DAP).At least 21 miRNAs were differentially expressed.The differential expression of three of these miRNAs,i.e.,miR528a,miR167a and miR160b,at each stage was verified by q RT-PCR.The results indicated that these miRNAs might be involved in kernel development.In addition,the predicted functions of target genes indicated that most of the target genes are involved in signal transduction and cell communication pathways,particularly the auxin signaling pathway.The expression of candidate germination-associated miRNAs was analyzed by hybridization to a maize genome microarray,and revealed differential expression of genes involved in plant hormone signaling pathways.This finding suggests that phytohormones play a critical role in the development of maize kernels.We found that in combination with other miRNAs,miR528a regulated a putative laccase,a Ring-H2 zinc finger protein and a MADS box-like protein,whereas miR167a and miR160b regulated multiple target genes, including ARF(auxin response factor),a member of the B3 transcription factor family.All three miRNAs are important for ear germination,development and physiology.The small RNA transcriptomes and mRNA obtained in this study will help us gain a betterunderstanding of the expression and function of small RNAs in the development of maize kernel.
Crown Copyright©2013 Production and hosting by Elsevier B.V.on behalf of Crop Science Society of China and Institute of Crop Science,CAAS.All rights reserved.
1.Introduction
Seed dormancy and germination are controlled by intrinsic hormonal and metabolic pathways,the components of which are influenced by external environmental cues[1–3].Germination is a key process that allows a seed embryo to grow and develop into a photosynthetic organism.The process of germination starts with the hydration of quiescent seed and ends with the onset of elongation of the embryo axis,which corresponds to the emergence of the radicle from the seed [4,5].Despite the impressive amount of information on the physiology of germination[4]and advances in understanding its molecular basis[5,6],recent transcriptome analyses have shown that dormancy in Arabidopsis(Arabidopsis thaliana)is associated with specific patterns of gene expression that are different from those in germinating seeds[7,8].Recently,a new paradigm of post-transcriptional gene regulation has evolved as a result of the discovery of hundreds of miRNAs in maize(Zea mays L.).The diverse expression patterns of miRNAs and the large number of potential target mRNAs suggest their involvement in the regulation of a variety of developmentally related genes atthe post-transcriptionallevel.Accumulating evidence indicates thatmiRNAs may function as ear germination suppressors during maize ear development and that they may have critical functions in growth,development,and responses to biotic and abiotic stresses.In plants,miRNAs regulate diverse genes and pathways such as those for development,hormone signaling,stress response and trans-acting siRNAs [9,10].Interestingly,phytohormones regulate plant development via a complex signal response network.Five major plant hormone genes are involved in the signaling pathway: auxin,cytokinin,gibberellin,abscisic acid,and ethylene. Many of the target genes associated with auxin are involved in ear development[1,11–13].This finding led to the hypothesis that miRNAs play an important role in regulation of target genes during ear germination.The key roles played by GA(gibberellins)and ABA(abscisic acid)in ear germination and early development have long been established[14].Furthermore,previous research determined that in viviparous(vp) mutants in maize and other cereal grains,the embryo fails to become dormant and undergoes precocious germination on the mother plant.
miRNAs are a class of small single-stranded non-coding RNAs ranging in length from 20 to 24 nucleotides(nt)[15,16]. MostmiRNAtargets are mRNAs ofprotein-coding genes,which, upon targeting,are cleaved or repressed at the translational level[16–19].Thus,miRNAs act as negative regulators of gene expression.In plants,most miRNAs regulate target gene expression via mRNA degradation[20].MiRNAs recognize completely or partially complementary sequences in their target mRNAs and guide them to cleavage or translational arrest.Plant miRNAs usually recognize one motif in the coding region of their targets and affect their stability.It is thought that better complementarity between plant miRNAs and their targets favors translational arrest rather than cleavage.The high degree of complementarity between plant miRNAs and their target mRNAs has allowed the identification of targets using algorithms that scan the genome for mRNA–miRNA complementarity[21].Refinements in this method have increased the reliability of predictions[22].When a miRNA targets multiple mRNAs,the targeted genes are often members ofa gene family,and miRNAs that are conserved between Arabidopsis and rice(Oryza sativa L.)also tend to have conserved targets[21]. Intriguingly,plant miRNAs display a striking propensity to target the mRNAs of transcription factors that have already been established as key developmental regulators or their closely related factors.A number of computational methods have been reported for the identification of plant miRNAs [23–26].Research on plants revealed that short sequences of mature miRNAs are conserved and exhibit high complementarity to their target mRNAs[24].Hence,candidate miRNAs can be detected using their conserved complementarities to target mRNA if the mRNA target sequence is known.Conversely,it has also been shown that the secondary structures of miRNA precursors(pre-miRNAs)are relatively more conserved than pri-miRNA sequences(the precursors of pre-miRNAs)[27].For instance,through sequence homology analysis,30 potential miRNAs were predicted in cotton(Gossypium spp.)[28],and an additional 58 miRNAs were identified in wheat(Triticum aestivum L.)[29].The majority of plant miRNAs studied to date are involved in regulating developmental processes[30,31]and they negatively regulate expression of their target genes at the post-transcriptional level.Computational methods for identifying miRNAs in plants are more rapid,less expensive,and easier than experimental procedures.However,these bioinformatics approaches can only identify miRNAs thatare conserved across organisms,and any computationally predicted miRNAs should also be confirmed via experimental methods.The direct cloning ofsmall RNAs fromplants is one of the basic approaches ofmiRNA discovery and has been used to isolate and clone small RNAs from various plant species such as Arabidopsis and rice [32–34].Many miRNAs are broadly expressed but can be detected only under certain environmental conditions,at different plant developmental stages,or in particular tissues.Therefore,plant samples from specific times,different tissues,and different stress conditions(biotic and abiotic stress-induced)are used for miRNA cloning.The most common plant species used for direct cloning are Arabidopsis[31,34,35],rice[36],cottonwood(Hibiscus tiliaceus)[37]and wheat[38].The most important advantage of cloning small RNAs compared to computational approaches is the opportunity to find non-conserved and species-specific miRNAs.Efficient and appropriate miRNA detection and quantification methods are essential for understanding the function of a given miRNA under different conditions or in different tissues.In this study,we constructed a small RNA library to represent the full complement of individual small RNAs and characterized miRNA expression profiles in pooled developing ears of maize(Z.mays L.).In addition,we carried out functionalpredictions of the target genes of candidate miRNAs.The small RNA transcriptomes and mRNAs obtained in the study will help us gain a better understanding of the expression and function of small RNAs in developing maize kernels.
2.Materials and methods
The inbred maize line A318 is a viviparous mutant developed from the tropical germplasm S37 by spaceflight breeding (Fig.1).Ears at 10,15,20,22,25 and 30 days after pollination (DAP)were collected from plants grown under standard greenhouse conditions.Kernels were dissected from the ears,immediately frozen in liquid nitrogen,and stored at−80°C until RNA extraction.
Isolated total RNA was size-fractionated on a 15%Tris–borate–EDTA(TBE)urea polyacrylamide gel to enrich molecules of 15–30 nt.The small RNA was ligated with adapters (5′-GTCTCTAGCCTGCAGGATCGATG-3′)and(5′-AAAGATCC TGCAGGTGCGTCA-3′),and(5′-GTCTCTAGCCTGCAGGATCG ATG-3′)and(5′-AAAGATCCTGCAGGTGCGTCA-3′)using T4 RNA ligase and size-fractionated on a 15%TBE urea polyacrylamide gel.The resultant RNA was reversely transcribed to cDNA with a small RNA RT-primer(5′-CAAGCAGAAGAC GGCATACGA-3′),and the cDNA was then directly subcloned into vector p MD18-T(TaKaRa).These tandem cDNA fragments were transformed into Escherichia coli strain DH5 by electroporation.Colony PCR was performed using 5′and 3′primers,and clones with lengths of 60–80 bp were used for sequencing according to the manufacturer's protocols(Colony PCR Made Easy,http://www.lucigen.com/colonyPCR).
Small RNAs(200 nt)were isolated with the mirVana PARIS Kit(Ambion)according to the manufacturer's instructions.For reverse transcription(RT),1 μg of small RNA was treated with the miScript Reverse Transcription Kit(Qiagen)at 37°C for 60 min and a final incubation at 95°C for 5 min.Real-time PCR of miRNAs was carried out using the miScript SYBR Green PCR kit(Qiagen)in an Applied Biosystems 7500 real-time PCR machine(ABI).PCR was conducted at 95°C for 15 min,followed by 40 cycles of incubation at 94°C for 15 s,55°C for 30 s,and then 70°C for 30 s.Each PCR was repeated at least three times. All samples were normalized to 5S rRNA expression and fold change expression was calculated according to the 2−ΔΔCtmethod as described previously[39].
High-quality small RNA reads larger than 18 nt were extracted from the raw reads and mapped to maize genome sequences(http://www.maizesequence.org)using SOAPaligner/ soap2(http://soap.genomics.org.cn/soapaligner.html)[40]. Matched sequences were then queried against non-coding RNAs from the Rfam database(http://www.sanger.ac.uk/ Software/Rfam)and the ncRNA database(http://www.ncrna. org/frnadb/blast/fRNAdb).Most non-miRNAs,non-siRNAs and mRNA degradation fragments were removed by a BLASTn search of the NCBIGenBank database(http://www.ncbi.nlm.nih. gov/blast/Blast.cgi)[41].Any small RNAs with exact matches to these sequences were excluded from further analysis.miRNAs were predicted with Mireap(https://sourceforge.net/projects/ mireap/).Secondary structures of the predicted miRNAs were confirmed using the RNAfold online tool(http://rna.tbi.univie. ac.at/cgi-bin/RNAfold.cgi).miRNA target candidates were predicted using the PatScan program[42]based on methods described by Lu[37],with penalty scores≤3 for mismatched patterns in the miRNA/mRNA duplexes.Putative target genes were manually selected from these candidates based on their location in the maize genome.Functions of the predicted target genes were assigned manually according to the functions of the best hits from the BLAST search[41,43]against the NCBI database(http://www.ncbi.nlm.nih.gov/blast/Blast.cgi).For the predicted novel miRNA sequences,conservation in other plant species was examined by searching the nucleotide databases with BLASTn[41]to identify their homologs and surrounding sequences.These germination-related miRNAs were also aligned with the maize genome using PatScan[42].To analyze whether the matched sequence could form a suitable hairpin,the sequences of candidate precursors were analyzed using RepeatMasker(http://www. repeatmasker.org/)to eliminate repetitive sequences.Sequences surrounding the matched sequence(100–200 nt to either side)were extracted and run through RNAfold(http://rna. tbi.univie.ac.at/cgi-bin/RNAfold.cgi).

Fig.1–S37(A)and its viviparous mutant A318(B)under normal conditions.
Most targets of miRNAs in plants have one miRNA-complementary site located in the coding region and occasionally in the 3′or 5′un-translated regions(UTRs)[21,36,38,44,45], and plant miRNAs exhibit perfect or near-perfect complementarity with their target mRNAs[46].We adopted a set of previously reported rules to predict miRNA targets[36,47]. These rules allow one mismatch in the region complementary to nucleotide positions 2 to 12 of the miRNA,do not allow a mismatch at position 10/11,which is a predicted cleavage site, and allow three additional mismatches between positions 12 and 22,but with no more than two continuous mismatches. Therefore,candidate miRNA target genes were determined using publicly available prediction algorithms,including miRU [48],the target search in WMD web[49],and the prediction tool in the UEA plant sRNA toolkit.These programs were used with their default settings.The microarrays used in this study were obtained from GSE9386,entitled“Genome-wide analysis of gene expression profiles during the kernel development of maize(Z.mays L.)”.The raw data from microarray hybridization was exported from GenePix suites 6.0(Axon,USA)and imported into LIMMA with annotation and spot types[50].Spots with a negative flag value were assigned a weighting of 0.1 in the subsequent analysis.Background-subtracted signal intensities were normalized using two-step normalization, consisting of print-tip group loss(within-array normalization)and between-array scale normalization.The adjusted p value was then assessed using the false discovery rate.To identify a statistically significant differentialexpression of genes, p=0.01 was used as a criterion.To obtain probe annotations, the consensus representative sequences of all probes were searched using BLAST against the TIGR rice protein database (http://www.tigr.org/),the TAIR Arabidopsis protein database (http://www.arabidopsis.org/),and the UniProt Knowledgebase (http://www.ebi.ac.uk/).Functional annotations were then assigned based on sequence similarity(with E-value of 10−5) with manual adjustment when necessary.All of the probes were grouped into functional categories and metabolic pathways based on the Mips Functional Catalogue(http://mips.gsf. de/)and KEGG[51–53].Adhering to the established method [54,55],we identified statistically enriched KEGG pathways and gene families of transcription factors in differentially expressed genes based on a background distribution from the whole chip.The expression levels of genes were measured by detection calls and signal intensities using Micro Array Suite 5.0 software with a target signal of 100.All pair-wise differentially expressed genes were identified using SAM software(http://www-stat.stanford.edu/~tibs/SAM/)to analyze data from all remaining maize probe sets.A false discovery rate parameter of 1%was set for the SAM analysis.Genes that were called absent more than twice among the three replicates in both the control and treatment arrays were then considered to be not unexpressed under both conditions and were excluded from the list.
A 5 μg sample of total RNA was used for cDNA synthesis using the Invitrogen Reverse Transcription Reagents Kit (Invitrogen).Gene-specific primers were designed using Primer Express 2.0 software(Applied Biosystems)and synthesized by Sangon Biotech Co.Ltd.(Shanghai).Relative quantitative analysis was performed using an Applied Biosystems 7900HT real-time PCR system(Applied Biosystems)under the following conditions: 94°Cfor3 min(1 cycle);94°Cfor 30 s,60°Cfor 30 s,and 72°Cfor 30 s(40 cycles).Transcript abundance was identified using SYBR Green PCR Master Mix(Applied Biosystems).Each reaction contained 1×buffer,0.25 μmol L−1of each primer,and approximately 2 ng ofcDNAin a finalvolume of 20 μL.Three replicates were employed for each tested sample and template-free negative control.Mitochondrial 5S RNA was used as an internal controlto normalize alldata.Melting curves were performed on the product to test whether only a single product was amplified withoutprimer-dimers and other bands.The resultantproducts with all primer combinations were initially visualized on 2% agarose gels to confirm the generation of a single product of the correct size.
3.Results
To identify miRNAs in developing kernel of a maize viviparous mutant at different developmental stages,RNAs of 18 to 26 nt in length were purified and cloned from small RNAs(~200 nt) isolated from germinating maize kernels for subsequent sequence analysis.Approximately 64 concatamer clones were sequenced to generate 540 sequences after discarding low-quality and self-ligated linker sequences.Of these,56 small RNA-cDNAsequences were outside the expected range of nucleotide lengths(18–26 nt).Only the nucleotide sequences that were longer than 18 nt were subjected to detailed analysis. The distribution of different lengths of nucleotide sequences found in this library is shown in Fig.2.
We categorized all identified sequences according to their properties using criteria reported elsewhere for different types of small RNAs.The 540 sequences identified in the library consisted of approximately 19.0%miRNA,13.0%mRNA,12.0% rRNA,9%tRNA,8.0%repeat-associated siRNA,5.7%small antisense RNA,6.0%tiny noncoding RNA,2.3%small nuclear RNA and 25.0%of sequences that had no matches in the maize genome.
In the cDNA library,a total of 108 sequences were found to be miRNA-like molecules.Twenty-six newly identified sequences perfectly matched the maize genome and were able to adopt hairpin structures.The lengths of these newly identified miRNAs ranged from 19 to 24 nt,and 10 of them began with a 5′uridine,a characteristic feature of miRNAs. Twenty-one of these miRNAs were reported in miRBase 12.0 for different species,including maize,16 were registered for other species,and 5 were new.
For each miRNA,the corresponding ear genomic DNA sequences and their locations were identified.The 5′or 3′flanking genomic sequences were then tested for ability to fold into miRNA precursor hairpin structures of approximately 70 nt using the Mfold web server[56].The presence of small RNA clones with the proper positioning within an arm of the hairpin suggested that they could have been excised during dicer processing in the cells.In nearly all of those cases,the sequences were found to be conserved in different species, including the predicted precursors.Moreover,5 miRNA families (i.e.,Zma-miR160,Zma-miR164,Zma-miR167,Zma-miR171 and Zma-miR528)were conserved in at least three species and 5 miRNAlociwere specific to the maize ear(Table 1).To determine whether our new miRNAs are conserved among closely relatedspecies,we searched for homology of their precursor sequences in the ENSEMBL genome databases.The results revealed that 16 precursor loci were conserved in at least six species.All of the newly cloned miRNAs were conserved as mature sequences in the genomes of different species.Thermo-dynamically stable hairpin structures were found for these new conserved miRNAs (Fig.3).
It was shown that plant miRNAs exhibit a high degree of sequence complementarity to their targets,allowing for effective target prediction[57].Target prediction analysis,therefore,was performed for the germination-related zma-miRNAs (Tables 2 to 4).
The expression patterns of three annotated miRNAs (i.e.,miR528a,miR167a and miR160b)at all six sampling times were analyzed using qRT-PCR(Fig.4).Because the small RNAs were cloned with a library derived from different times of maize ear development,they were able to resolve the expression profiles of the new miRNAs.Because of the distinct nature and function of germination,the cells and tissue compartments in the ear showed continuous change throughoutthe process of development.
We sought to identify the major biological processes and signaling pathways that are most likely affected by a group of miRNAs during development of the maize ear.Several potential targetgenes were predicted to be associated with cloned miRNAs based on our filtering and screening procedures.The miRNAs encoding proteins involved in regulation of mating were represented at higher frequencies in our library.Furthermore,detailed gene ontology(GO)analysis showed that screened sub-sets of miRNA target genes were associated with ear development, function,and regulation(Fig.5)involved in primary and secondary metabolism,signal transduction,transcription and regulation,and protein processing and destination
To identify the target genes associated with ear germination identified in our research,identical genes were extracted from GSE9386 raw data.We detected 92 differentially expressed genes (P=0.05)associated with germination during the process of ear development.Most of the changes in differentially expressed genes were observed between 10 and 15 DAP,while 23 genes had significant fold changes between 25 and 35 DAP.To elucidate the expression profiles of the differentially expressed genes, we transformed comparisons of two consecutive time points into a comparison using expression levels at 10 DAP as a common reference.A selection of differentially expressed genes associated with the candidate miRNA in ear germination listed in Table 5 includes genes related to cell division, starch metabolism,storage proteins,and hormone signaling pathways.Both up-and down-regulation occurred during the ear germination process.
Down-regulated gene expression predominated during the periods 15 to 25 and 25 to 35 DAP,whereas up-regulated gene expression predominated from 10 to 15 DAP.Thus,we added 20 and 22 DAP,which lie within the period from 15 to 25 DAP, and 30 DAP,which lies between the 25 and 35 DAP,to study the mechanism in more detail.The genes related to the ABA signaling pathway(e.g.,serine/threonine protein kinase and transcription factor MYB30)and the gibberellin(GA)signaling pathway were also included in these two clusters.These genes were associated with ear germination and the candidate miRNAs.Through analysis of gene expression patterns, we found that these genes may be involved in the entire germination process in the maize ear,and we concluded that miR167a/miR160b and miR528a might play major roles in ear germination by modifying their target genes,in combination with other miRNAs(Fig.6).
To confirm the accuracy and reproducibility of the microarray results,real-time PCR was carried out using 8 differentially expressed genes associated with miR167a/160b and miR528a(Table 6).We added 20 DAP and 22 DAP in the period between 15 and 25 DAP and 30 DAP between 25 and 35 DAP to study the detailed mechanism by real-time PCR.The results showed that the expression patterns of these genes were well correlated with the results from microarray analysis.However, given the wider dynamic range and greater sensitivity of realtime PCR,the variation of differentially expressed genes from real-time PCR was more significant than that from the microarray analysis;we found that 22 DAP is an important turning point in ear germination.
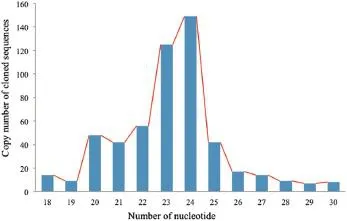
Fig.2–Size distribution of 540 small RNA sequences cloned from maize kernels.

Table1–ComparisonofnumbersofmiRNAfamily members in maize,rice,Arabidopsis and soybean.
4.Discussion
MicroRNAs play integral roles in gene regulatory networks as one of the most abundant classes of gene regulators.The expression and activity of plant miRNAs can be regulated in many ways,including transcriptional control,as well as regulation imposed at the levels of miRNA processing and action. Moreover,changes in the expression of even a single miRNA could have a significant impact on the outcome of diverse cellular activities regulated by the product of that mRNA. Repression of the target transcript by miRNAs may occurthrough translational inhibition,accelerated exonucleolytic mRNA decay,or slice within miRNA–mRNA base pairing[58]. Beyond the strict conservation of miRNAs across different species,some miRNAs appear to be species-specific.Compared with computational or heterologous approaches,direct miRNA cloning has the advantage of identifying non-conserved and new miRNAs.
There are a number of highly conserved miRNA families in maize.In the present study,cloning and expression analysis led to the identification of 26 miRNA variants belonging to 21 miRNAfamilies,as wellas 5 new miRNAs and 16 putatively new miRNAs.Non-conserved plant miRNAs presumably emerge and dissipate in short evolutionary time scales.Representation of many known and novel miRNAs in this single library indicates the presence of miRNAs that are not yet discovered. The identification of a large number of miRNAs that are not previously reported in maize,at least 10 of which are conserved in monocots,suggests that many more monocotor maize-specific miRNAs are yet to be identified.Certainly, additional tissues should be evaluated for further discovery of miRNAs in maize,coupled with similar studies in related monocots.This will help establish how many of the currently maize-specific miRNAs are conserved in other monocotspecies. Moreover,both in-depth analysis of the existing library andorgan-specific analysis of individual miRNAs will give insights into the functional mechanisms and pathways involved in particular in ear germination and ear development in general.
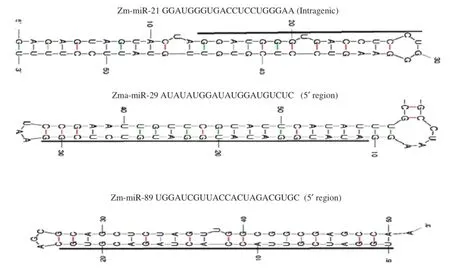
Fig.3–Predicted secondary structure of three selected miRNA hairpins.
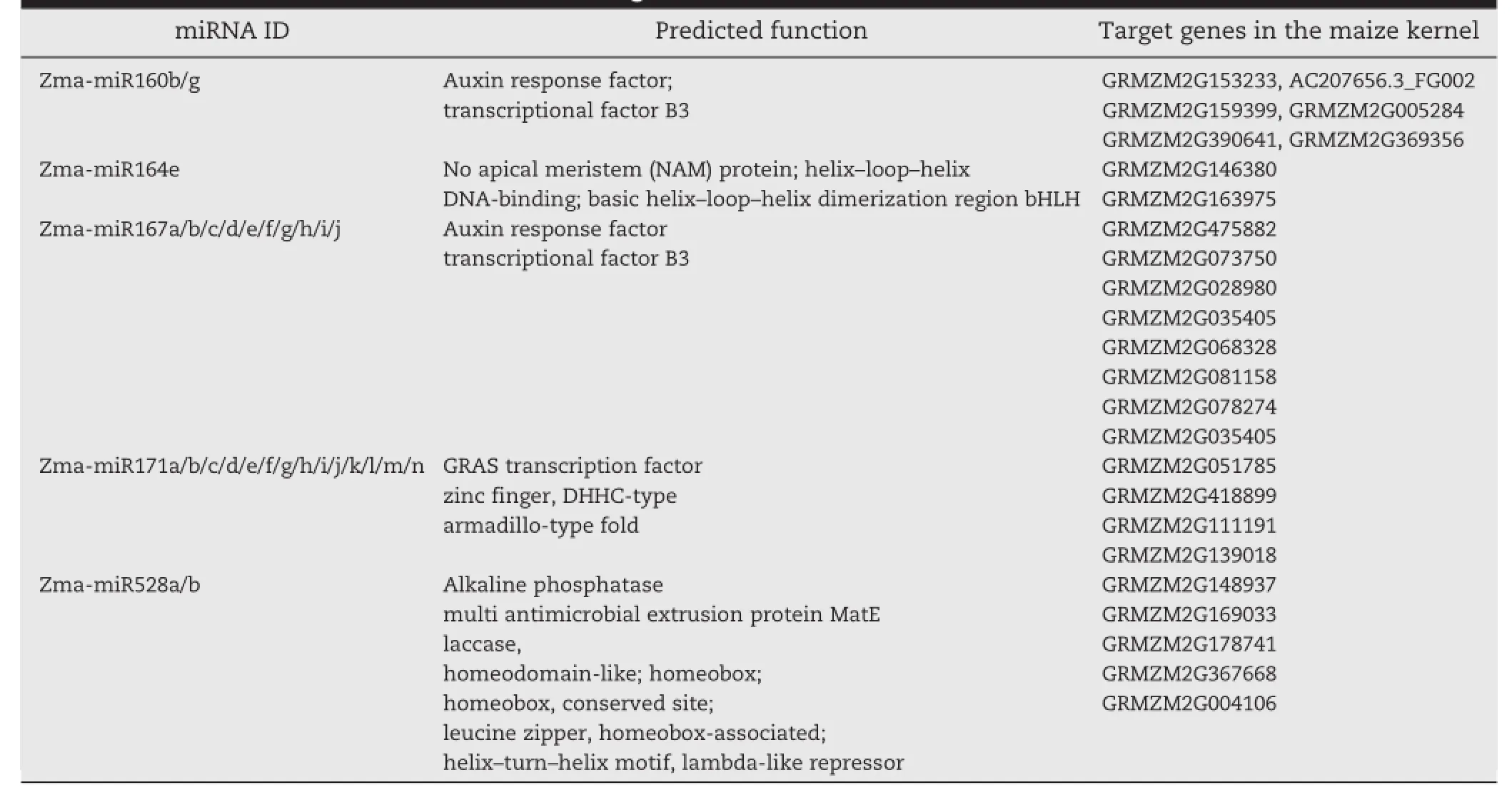
Table 2–Non-conserved miRNAs in maize kernel germination.
Recent studies have demonstrated that miRNAs in Arabidopsis,rice,and other plant species target transcripts that encode proteins involved in diverse physiological processes by predominantly targeting transcription factors[25,37,44,59].In this study,we predicted 90 unigenes as putative miRNA targets in maize ears,with one-third of the predicted targets of miRNAs being mRNAs of transcription factors,including AUX_IAA,MYB,ARF,bZIP,b HLH and MADS.Other target genes include those encoding putative NADH dehydrogenase (ubiquinone oxidoreductase),a putative laccase,ethyleneresponsive factor-like protein 1,pyruvate,phosphate dikinase, a chloroplast precursor,thymidine kinase,a cytochrome P450-like protein,and a transposase,suggesting that miRNAs in maize ears are involved in a broad range of physiological functions.Further analysis indicated that the targets of 16 conserved miRNAs from maize ears are also conserved among other plant species,implying that conserved miRNAs serve conserved biologicalroles.Moreover,these targets were distinct fromtheir Arabidopsis and rice homologs(especially the targets of the non-conserved miRNAs),indicating that they may be involved in ear-specific processes in maize.Itwill be interesting to identify the functions of these predicted target genes in maize.
Most target mRNAs of plant miRNAs have only a single miRNA-complementary site located in the coding regions or occasionally in the 3′or 5′UTR[21,25,44,60].Consistent with these reports,maize ear miRNAs are predicted to target coding regions.Although 3′UTRs were predicted to be target sites for plant miRNAs in only a few previously reported cases, 3 of the 16 targets of novel maize miRNAs reported in this study had target sites within the 3′UTR,four were within a coding region,and 9 were in the 5′UTR.This bias might reflect a mechanistic preference for translational repression.The fate of an mRNA may depend on the degree of complementarity between a miRNA and its target mRNA;it appears that perfectly base-paired miRNAs mediate cleavage,whereas imperfectly base-paired miRNAs mediate translation repression[61].We found that half of the miRNAs targeting 5′UTRs were perfectly base-paired,indicating that they might cleave their target mRNAs to down-regulate expression.Future experiments will reveal whether these target genes are destined for degradation or translational repression.

Table 3–Conserved miRNAs in maize kernel germination.

Table 4–Novel miRNAs in maize kernel germination.
Phytohormones regulate plant development via a complex signal response network,especially auxin,cytokinin,gibberellin,abscisic acid,and ethylene.In our study,15 differentially expressed genes were involved in the auxin-signaling pathway in the course of the total developmental process(Table 2). MiR167 and miR160 were down-regulated after 22 DAP in developing viviparous kernels,implying that these miRNAs might be involved in receiving a phytohormone signal during the final stages of ear development.Auxin-responsive factor genes ARF3 and ARF6b were predicted to be targets of zmamiR167 and miR160.However,ARF3 and ARF6b were upregulated after 22 DAP by microarray hybridization,and variation of differentially expressed genes from real-time PCR was more significantthan that observed in the microarray analysis.Auxin response is regulated by various positive and negative feedback mechanisms during plant growth.ARFs regulate the transcription of early auxin-responsive genes,including AUX/IAA genes [62],whereas AUX/IAA proteins interact with ARFs and repress their activities[63].Auxin induces the targeted ubiquitination/ degradation of specific AUX/IAA proteins[64]and frees ARFs from repression by AUX/IAA proteins.ARFs are bound to AUX/IAA negative regulators,thus maintaining the ARFs in an inactive state.The binding of auxin to TIR1-related F-box proteins enhances AUX/IAA destruction via the proteasome, liberating ARF activity[65–67].We also found that the accumulation of ARF transcripts resulting from down-regulation of miR167/miR160 might enhance auxin response and thus enhance maize germination.Moreover,Liu et al.[68]reported that the regulation of ARF10 mRNA stability by the miR160 miRNA implicated ARF10 in modulating ABA responsiveness during ear germination.More recently,it was shown that miR167 and miR160 are also regulated by ABA in rice,suggesting that they may also be involved in plant growth[69].ABA down-regulation ofmiR167,which regulates auxin response factor 3(ARF3)mRNA, suggests that ABA may cause increased ARF3 mRNA accumulation or translational promotion.Because ARF3 is a positive regulator in both female and male reproductive functions[70,71], the accumulation of ARF3 by alleviating miR167/miR160 regulation would lead to earlier female and male development and, consequently,earlier plant maturation.We also deduced that miR167 may interact with miR160 via the common target genes to promote maize ear development.Our study elucidated the importance of the auxin-signaling pathway in ear development in maize.The results point to a role of auxin in germinationassociated pathways and suggest that the interactions betweenboth auxin and ABA signaling pathways may contribute to the germination potentialof seeds.An analysis ofthe function ofkey components of auxin signaling in relation to after-ripening, germination potential,and vigor may revealnovelroles for auxin in these processes.However,further research is warranted to elucidate the interactions ofthese pathways in ear development.
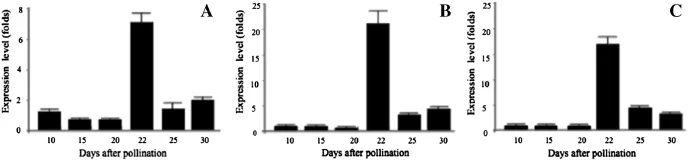
Fig.4–Detection and expression of candidate miRNAs miR167a(A),miR160b(B),and miR528a(C)at six developmental stages (days after pollination).
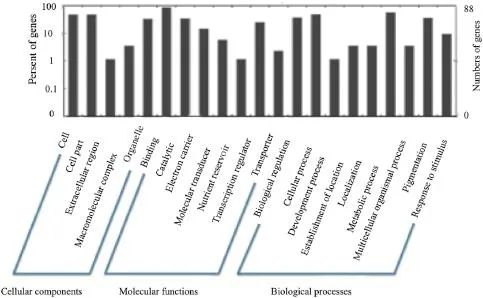
Fig.5–Detailed gene ontologies(GO)of screened total and sub-sets of miRNA target genes.
Among the differentially expressed transcription factors related to the candidate miRNAs in maize ear germination (Table 3),there are 3 bZIP transcription factors,which regulate the expression of zeins.A gene encoding Ring-H2 zinc finger protein MZ00003207,which was up-regulated from 22 to 30 DAP, may mediate auxin-and salicylic-acid-inducible transcription [72].Furthermore,the MADS box-like protein MZ00022813, which had the lowest expression at 22 DAP,may bind to the promoters of genes regulated by multiple stimuli,such as light and hormones[73,74].At 22 DAP,miR528 was up-regulated in maize germination,indicating that these miRNAs might be involved in receiving phytohormone signals.The homeobox– leucine zipper family protein MZ00030111,a Ring-H2 zinc finger protein,a MADS box-like protein,and the putative laccase MZ00049071 were predicted as the targets of zma-miR528.It was concluded,therefore,that the bZIP transcription factors might play a significant role in kernel filling and response to hormones.In our study,a gene encoding ARF was up-regulated during kerneldevelopment,suggesting thatit also plays a similar role in differentiation during maize embryogenesis.Moreover, many putative protein kinase genes were differentially expressed at various times,which were involved in signaling transduction pathway during maize ear development.For example,homeobox–leucine zipper family protein,a member of the LRR(leucine-rich repeat family protein)subfamily,might be required to increase cell size and the rate of embryonic development.A gene encoding a homeobox–leucine zipper family protein was up-regulated from 15 to 25 DAP,suggesting that this kinase might function in forming organs in the maize embryo.Therefore,we deduced that the accumulation of bZIPtranscripts,MADS box-like proteins,and putative laccase resulting from the down-regulation of miR528 might enhance auxin response and,in turn,seed germination in the final stage of seed development(after 22 DAP).However,further work is required to elucidate the functions of these protein kinases.
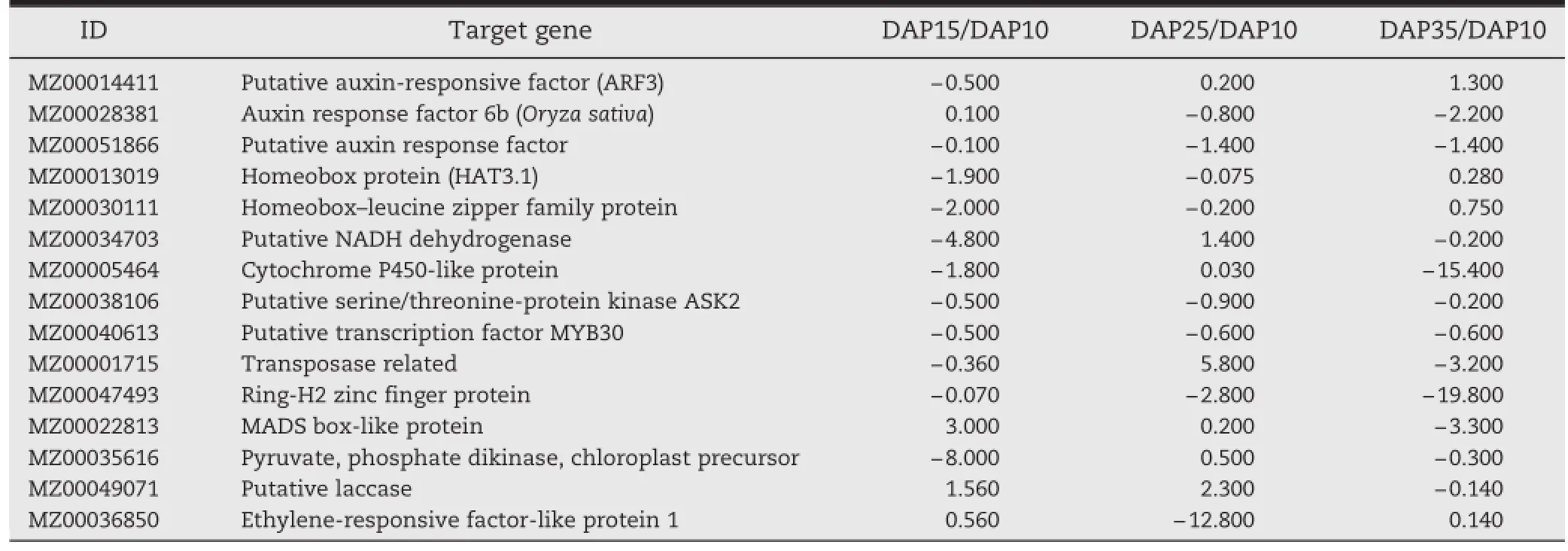
Table 5–Differential expression of genes related to candidate miRNAs(miR167a/160b and miR528a)in maize ear germination.

Fig.6–Patterns of gene expression in developing maize kernels.The blue line denotes all the candidate genes in developing maize kernels.The red line denotes the entire candidate genes based on the whole gene set of ZmB73.
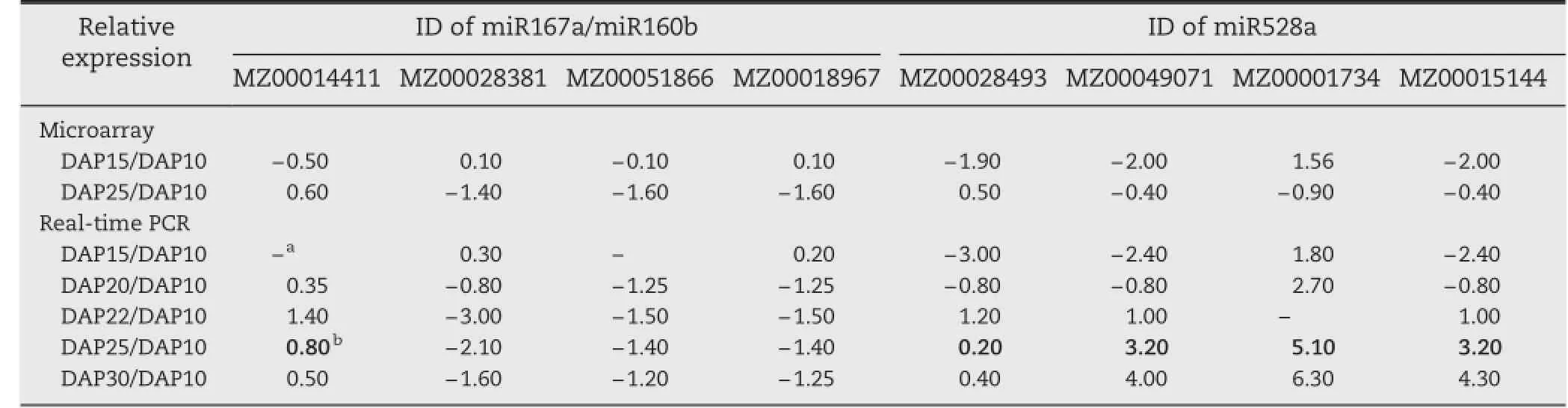
Table 6–Validation of microarray results by real-time PCR assay of differentially expressed genes associated with miR167a/ miR160b and miR528a.
5.Conclusions
By constructing a small RNA library and characterizing miRNA expression profiles in pooled maize ears at 10,15,20,22,25 and 30 DAP,at least 21 miRNAs were differentially expressed. qRT-PCR verification for miR528a and miR167a/miR160b indicated that these miRNAs might be involved in ear development and germination.In addition,functional predictions of target genes indicated that most of these differentially expressed miRNAs tended to have target genes that were involved in signal transduction and cell communication,particularly those involved in the auxin-signaling pathway.The results of gene expression analysis of candidate germination-associated miRNAs performed by microarray hybridization with a maize genome array demonstrated the differential expression of genes involved in plant hormone signaling pathways.This suggested that phytohormones might play a critical role in the maize ear developmental process.We showed that in combination with other miRNAs,miR528a regulates a putative laccase,a Ring-H2 zinc finger proteinand a MADS box-like protein,whereas miR167a and miR160b regulate target genes including ARF(auxin response factor),a member of the B3 transcription factor family that is important for ear germination and physiology.Thus the small RNA transcriptomes and mRNA obtained in this study provide considerable insight into the expression and function of small RNAs in the development of viviparous kernels.
Acknowledgments
This study was supported by grants from the Educational Commission of Sichuan Province(No.2006J13-039),the Doctoral Program Foundation of Institutions of Higher Education of China (No.20095103120002),and the National Natural Science Foundation of China(No.30900901).
R E F E R E N C E S
[1]M.J.Holdsworth,L.Bentsink,W.J.Soppe,Molecular networks regulating Arabidopsis seed maturation, after-ripening, dormancy and germination,New Phytol.179(2008)33–54.
[2]W.E.Finch-Savage,G.Leubner-Metzger,Seed dormancy and the control of germination,New Phytol.171(2006) 501–523.
[3]B.Kucera,M.A.Cohn,G.Leubner-Metzger,Plant hormone interactions during seed dormancy release and germination, Seed Sci.Res.15(2005)281–307.
[4]A.A.Khan,The Physiology and Biochemistry of Seed Development,Dormancy and Germination,Elsevier Biomedical Press,New York,1982.
[5]J.Bove,M.Jullien,P.Grappin,Functional genomics in the study of seed germination,Genome Biol.(2001)3, (reviews1002.1001-reviews1002.1005).
[6]B.J.W.Dekkers,S.Smeekens,Sugar and abscisic acid regulation of germination and transition to seedling growth, in:K.Bradford,H.Nonogaki(Eds.),Seed Development, Dormancy and Germination,Annu Plant Rev,27,2007, pp.305–327.
[7]C.S.Cadman,P.E.Toorop,H.W.Hilhorst,W.E.Finch-Savage, Gene expression profiles of Arabidopsis Cvi seeds during dormancy cycling indicate a common underlying dormancy control mechanism,Plant J.46(2006)805–822.
[8]E.Carrera,T.Holman,A.Medhurst,D.Dietrich,S.Footitt, Seed after-ripening is a discrete developmental pathway associated with specific gene networks in Arabidopsis,Plant J. 53(2008)214–224.
[9]E.Allen,Z.Xie,A.M.Gustafson,J.C.Carrington, microRNA-directed phasing during trans-acting siRNA biogenesis in plants,Cell 121(2008)207–221.
[10]E.J.Sontheimer,R.W.Carthew,Silence from within:endogenous siRNAs and miRNAs,Cell 122(2005)9–12.
[11]X.Fu,N.P.Harberd,Auxin promotes Arabidopsis root growth by modulating gibberellin response,Nature 421 (2003)740–743.
[12]M.Ogawa,A.Hanada,Y.Yamauchi,A.Kuwahara,Y.Kamiya, Gibberellin biosynthesis and response during Arabidopsis seed germination,Plant Cell 15(2003)1591–1604.
[13]R.L.Young,A.V.Badyaev,Evolution of ontogeny:linking epigenetic remodeling and genetic adaptation in skeletal structures,Integr.Comp.Biol.47(2007)234–244.
[14]E.Nambara,A.Marion-Poll,Abscisic acid biosynthesis and catabolism,Annu.Rev.Plant Biol.56(2005)165–185.
[15]B.Bartel,D.P.Bartel,MicroRNAs:at the root of plant development?Plant Physiol.132(2003)709–717.
[16]D.P.Bartel,MicroRNAs:genomics,biogenesis,mechanism, and function,Cell 116(2004)281–297.
[17]M.J.Aukerman,H.Sakai,Regulation of flowering time and floral organ identity by a microRNA and its APETALA2-like target genes,Plant Cell 15(2003)2730–2741.
[18]P.Brodersen,L.Sakvarelidze-Achard,M.Bruun-Rasmussen, P.Dunoyer,Y.Y.Yamamoto,Widespread translational inhibition by plant miRNAs and siRNAs,Science 320(2008) 1185–1190.
[19]M.W.Jones-Rhoades,D.P.Bartel,B.Bartel,MicroRNAs and their regulatory roles in plants,Annu.Rev.Plant Biol.57 (2006)19–53.
[20]D.V.Dugas,B.Bartel,MicroRNA regulation of gene expression in plants,Curr.Opin.Plant Biol.7(2004)512–520.
[21]M.W.Rhoades,B.J.Reinhart,L.P.Lim,C.B.Burge,B.Bartel, Prediction of plant microRNA targets,Cell 110(2002)513–520.
[22]M.W.Jones-Rhoades,D.P.Bartel,Computational identification of plant microRNAs and their targets,including a stress-induced miRNA,Mol.Cell 14(2004)787–799.
[23]I.M.Ehrenreich,M.Purugganan,MicroRNAs in plants:possible contributions to phenotypic diversity,Plant Signal.Behav.3 (2008)829–830.
[24]P.Laufs,MicroRNA regulation of the CUC genes is required for boundary size control in Arabidopsis meristems,Development 131(2004)4311–4322.
[25]A.Adai,C.Johnson,S.Mlotshwa,S.Archer-Evans,V. Manocha,Computational prediction of miRNAs in Arabidopsis thaliana,Genome Res.15(2005)78–91.
[26]X.Chen,Y.Ba,L.Ma,X.Cai,Y.Yin,Characterization of microRNAs in serum:a novel class of biomarkers for diagnosis of cancer and other diseases,Cell Res.18(2008) 997–1006.
[27]R.Chatterjee,K.Chaudhuri,An approach for the identification of microRNA with an application to Anopheles gambiae,Acta Biochim.Pol.53(2006)303–309.
[28]M.Pang,A.W.Woodward,V.Agarwal,X.Guan,M.Ha, Genome-wide analysis reveals rapid and dynamic changes in miRNA and siRNA sequence and expression during ovule and fiber development in allotetraploid cotton(Gossypium hirsutum L.),Genome Biol.10(2009)R122.
[29]Y.Yao,G.Guo,Z.Ni,R.Sunkar,J.Du,Cloning and characterization of microRNAs from wheat(Triticum aestivum L.),Genome Biol.8(2007)R96.
[30]A.Y.Guo,Q.H.Zhu,X.Gu,S.Ge,J.Yang,Genome-wide identification and evolutionary analysis of the plant specific SBP-box transcription factor family,Gene 418(2008)1–8.
[31]A.C.Mallory,H.Vaucheret,Functions of microRNAs and related small RNAs in plants,Nat.Genet.38(2006)S31–S36.
[32]A.C.Mallory,D.V.Dugas,D.P.Bartel,B.Bartel,MicroRNA regulation of NAC-domain targets is required for proper formation and separation of adjacent embryonic,vegetative, and floral organs,Curr.Biol.14(2004)1035–1046.
[33]A.C.Mallory,MicroRNA-directed regulation of Arabidopsis AUXINRESPONSE FACTOR17 is essentialforproperdevelopment and modulates expression of early auxin response genes,Plant Cell 17(2005)1360–1375.
[34]C.Llave,Z.Xie,K.D.Kasschau,J.C.Carrington,Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA,Science 297(2002)2053–2056.
[35]C.Llave,K.D.Kasschau,M.A.Rector,J.C.Carrington,Endogenous and silencing-associated small RNAs in plants,Plant Cell 14 (2002)1605–1619.
[36]R.Sunkar,T.Girke,J.K.Zhu,Identification and characterization of endogenous small interfering RNAs from rice,Nucleic Acids Res.33(2005)4443–4454.
[37]X.Jian,Identification of novel stress-regulated microRNAs from Oryza sativa L,Genomics 95(2010)47–55.
[38]B.Zhang,Q.Wang,K.Wang,X.Pan,F.Liu,Identification of cotton microRNAs and their targets,Gene 397(2007) 26–37.
[39]M.W.Pfaffl,A new mathematical model for relative quantification in real-time RT-PCR,Nucleic Acids Res.29 (2001)e45.
[40]R.Li,Y.Li,K.Kristiansen,J.Wang,SOAP:short oligonucleotide alignment program,Bioinformatics 24(2008)713–714.
[41]S.F.Altschul,W.Gish,W.Miller,E.W.Myers,D.J.Lipman,Basic local alignment search tool,J.Mol.Biol.215(1990)403–410.
[42]M.Dsouza,N.Larsen,R.Overbeek,Searching for patterns in genomic data,Trends Genet.13(1997)497–498.
[43]S.F.Altschul,T.L.Madden,A.A.Schaffer,J.Zhang,Z.Zhang, Gapped BLAST and PSI-BLAST:a new generation of protein database search programs,Nucleic Acids Res.25(1997) 3389–3402.
[44]E.Bonnet,J.Wuyts,P.Rouze,Y.van de Peer,Detection of 91 potential conserved plant microRNAs in Arabidopsis thaliana and Oryza sativa identifies important target genes,Proc.Natl. Acad.Sci.U.S.A.101(2004)11511–11516.
[45]E.Bonnet,Y.He,K.Billiau,Y.van de Peer,TAPIR,a web server for the prediction of plant microRNA targets,including target mimics,Bioinformatics 26(2010)1566–1568.
[46]C.A.Kidner,R.A.Martienssen,The developmental role of microRNA in plants,Curr.Opin.Plant Biol.8(2005)38–44.
[47]R.Schwab,J.F.Palatnik,M.Riester,C.Schommer,M.Schmid, Specific effects of microRNAs on the plant transcriptome, Dev.Cell 8(2005)517–527.
[48]Y.Zhang,miRU:an automated plant miRNA target prediction server,Nucleic Acids Res.33(2005)W701–W704.
[49]S.Ossowski,R.Schwab,D.Weigel,Gene silencing in plants using artificial microRNAs and other small RNAs,Plant J.53 (2008)674–690.
[50]M.H.Han,S.Goud,L.Song,N.Fedoroff,The Arabidopsis double-stranded RNA-binding protein HYL1 plays a role in microRNA-mediated gene regulation,Proc.Natl.Acad.Sci. U.S.A.101(2004)1093–1098.
[51]E.Altermann,T.R.Klaenhammer,PathwayVoyager:pathway mapping using the Kyoto Encyclopedia of Genes and Genomes (KEGG)database,BMC Genomics 6(2005)60.
[52]M.Kanehisa,S.Goto,KEGG:Kyoto Encyclopedia of Genes and Genomes,Nucleic Acids Res.28(2000)27–30.
[53]H.Ogata,S.Goto,K.Sato,W.Fujibuchi,H.Bono,KEGG:Kyoto Encyclopedia of Genes and Genomes,Nucleic Acids Res.27 (1999)29–34.
[54]X.Mao,Automated genome annotation and pathway identification using the KEGG Orthology(KO)as a controlled vocabulary,Bioinformatics 21(2005)3787–3793.
[55]Y.H.Shi,Transcriptome profiling,molecular biological,and physiological studies reveal a major role for ethylene in cotton fiber cell elongation,Plant Cell 18(2006)651–664.
[56]M.Zuker,Mfold web server for nucleic acid folding and hybridization prediction,Nucleic Acids Res.31(2003)3406–3415.
[57]V.Mandhan,J.Kaur,K.Singh,smRNAome profiling to identify conserved and novel microRNAs in Stevia rebaudiana Bertoni,BMC Plant Biol.12(2012)197.
[58]A.Eulalio,E.Huntzinger,E.Izaurralde,Getting to the root of miRNA-mediated gene silencing,Cell 132(2008)9–14.
[59]S.Lu,Y.H.Sun,R.Shi,C.Clark,L.Li,Novel and mechanical stress-responsive microRNAs in Populus trichocarpa that are absent from Arabidopsis,Plant Cell 17(2005)2186–2203.
[60]R.Sunkar,J.K.Zhu,Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis,Plant Cell 16(2004) 2001–2019.
[61]C.H.de Moor,H.Meijer,S.Lissenden,Mechanisms of translational control by the 3′UTR in development and differentiation,Semin.Cell Dev.Biol.16(2005)49–58.
[62]T.Ulmasov,J.Murfett,G.Hagen,T.J.Guilfoyle,Aux/IAA proteins repress expression of reporter genes containingnatural and highly active synthetic auxin response elements, Plant Cell 9(1997)1963–1971.
[63]S.B.Tiwari,G.Hagen,T.Guilfoyle,The roles of auxin response factor domains in auxin-responsive transcription, Plant Cell 15(2001)533–543.
[64]A.M.Snijders,N.Nowak,R.Segraves,S.Blackwood,N.Brown, Assembly of microarrays for genome-wide measurement of DNA copy number by CGH,Nat.Genet.29(2001)263–264.
[65]T.Paciorek,J.Friml,Auxin signaling,J.Cell Sci.119(2006) 1199–1202.
[66]M.Quint,W.M.Gray,Auxin signaling,Curr.Opin.Plant Biol.9 (2006)448–453.
[67]W.D.Teale,I.A.Paponov,K.Palme,Auxin in action:signalling, transport and the control of plant growth and development, Nat.Rev.Mol.Cell Biol.7(2006)847–859.
[68]P.P.Liu,T.A.Montgomery,N.Fahlgren,K.D.Kasschau,H. Nonogaki,Repression of AUXIN RESPONSE FACTOR10 by microRNA160 is critical for seed germination and post-germination stages,Plant J.52(2007)133–146.
[69]Q.Liu,Y.C.Zhang,C.Y.Wang,Y.C.Luo,Q.J.Huang, Expression analysis of phytohormone-regulated microRNAs in rice,implying their regulation roles in plant hormone signaling,FEBS Lett.583(2009)723–728.
[70]P.Ru,L.Xu,H.Ma,H.Huang,Plant fertility defects induced by the enhanced expression of microRNA167,Cell Res.16(2006) 457–465.
[71]M.F.Wu,Q.Tian,J.W.Reed,Arabidopsis microRNA167 controls patterns of ARF6 and ARF8 expression,and regulates both female and male reproduction,Development 133(2006) 4211–4218.
[72]R.Niggeweg,C.Thurow,R.Weigel,U.Pfitzner,C.Gatz, Tobacco TGA factors differ with respect to interaction with NPR1,activation potential and DNA-binding properties,Plant Mol.Biol.42(2000)775–788.
[73]U.Schindler,A.E.Menkens,H.Beckmann,J.R.Ecker,A.R. Cashmore,Heterodimerization between light-regulated and ubiquitously expressed Arabidopsis GBF bZIP proteins,EMBO J. 11(1992)1261–1273.
[74]K.Boutilier,R.Offringa,V.K.Sharma,H.Kieft,T.Ouellet, Ectopic expression of BABY BOOM triggers a conversion from vegetative to embryonic growth,Plant Cell 14(2002) 1737–1749.
*Corresponding authors.Tel.:+86 835 2882714;fax:+86 835 2882154.
E-mail addresses:pangt1956@163.com(G.Pan),zzmmaize@gmail.com(Z.Zhang).
1These authors contributed equally to this work.
Peer review under responsibility of Crop Science Society of China and Institute of Crop Science,CAAS.
Microarray hybridization
qRT-PCR
Zea mays

- The Crop Journal的其它文章
- QTL mapping of starch granule size in common wheat using recombinant inbred lines derived from a PH82-2/Neixiang 188 cross
- Enhanced tolerance to drought in transgenic rice plants overexpressing C4photosynthesis enzymes
- Molecular approaches unravel the mechanism of acid soil tolerance in plants
- Enhanced tolerance to drought in transgenic rice plants overexpressing C4 photosynthesis enzymes
- HrcQ is necessary for Xanthomonas oryzae pv.oryzae HR-induction in non-host tobacco and pathogenicity in host rice
- Isolation and characterization of an isoamylase gene from rye