
Optimization of the Purification Methods for Recovery of Recombinant Growth Hormone from Paralichthys olivaceus
2013-04-17 10:05:56ZANGXiaonanZHANGXuechengMUXiaoshengandLIUBin
ZANG Xiaonan,ZHANG Xuecheng,MU Xiaosheng,and LIU Bin
1) Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences,Qingdao 266071,P. R. China
2) College of Marine Life Sciences, Ocean University of China, Qingdao 266003, P. R. China
1 Introduction
Paralichthys olivaceus,also called olive flounder,is one of the most important fish species in Chinese coastal fisheries.It is characterized by delicious and nutritious meat,low fat and calories,and high contents of protein and vitamins.With rapid development of flounder fishery,how to improve the quality and quantity of flounder has been of concern to relevant scientists and fish farmers.
Growth hormone (GH) is a protein hormone consisting of 170–190 amino acids,which participates in the controlling of several complex physiological processes in the animal body,e.g.,growth and metabolism (Knobil and Hotchkiss,1964).The amount of native GHs is extremely low in fish body,thus limiting its availability in research such as enzyme-linked immunosorbent assay (ELISA)and radioimmunoassay (RIA).At present,it is critical to obtain large-scale fish GH for study of fish physiology.The recombinant GH (rGH),which has the same function as native GH,is considered to have great practical value and positive impacts on fish farming (Liet al.,2003).Several studies have demonstrated that addition of rGH to the feed accelerates the growth of fish (Acostaet al.,2007; Liuet al.,2007).
An inexpensive and efficient way to obtain fish GH is to express this substance in recombinantEscherichia coli.To date,rGH of many fish species has been expressed in theE.colisystem,including salmon (Sekineet al.,1985),dolphin (Paduelet al.,1999),carp (Venugopalet al.,2002),and goldfish (Chanet al.,2003).Flounder rGH(r-fGH) was firstly expressed inE.coliin 1992,but its expression level relative to total cellular protein was only 8% (Watahikiet al.,1992).In 1998,the use of bicistronic construction in the expression plasmid substantially improved the yield of r-fGH,accounting for over 40% of theE.colicellular protein (Jehet al.,1998).However,the r-fGH was previously overexpressed inE.coliin the form of inclusion body.As the recovery of inclusion body largely dependeds on multiple factors such as characteristics of protein,pH,and oxidation/deoxidization system,the recovery rate and bioactivity of obtained r-fGH have been substantially low (Mukhopadhyay,1997).
In this study,several purification methods were tested and compared in order to identify a simple and efficient way for large-scale production of r-fGH with good receptor binding activity.The optimized r-fGH purification method would contribute to flounder farming.
2 Materials and Methods
2.1 Total RNA Extraction
Three 6-month old,100–120 g flounders (Paralichthys olivaceus) were provided by the experimental farm of the Yellow Sea Fisheries Research Institute,Chinese Acad-emy of Fishery Sciences,and temporarily sustained in the laboratory.The pituitary was dissected and immediately frozen in liquid nitrogen.Total RNA was extracted from the pituitary using EZ-10 Spin Column RNA Purification Kit (BBI,Canada) according to manufacturer’s instructions.To check RNA integrity,total RNA was analyzed by 1% agarose gel electrophoresis.The RNA extract showing obvious 18S rRNA and 28S rRNA bands on the agarose gel was considered intact and stored at −70℃.
2.2 Reverse-Transcription PCR of fGH cDNA
For amplification of fGH-coding genes,primers were designed based on the conserved regions of GH cDNA of three species ofPleuronectiformesdeposited in Genbank,i.e.,P.olivaceus(No.M23439),Verasper variegates(No.AF086787)and Hippoglossus hippoglossus(No.AB079-553).Primers sequences were as follows:
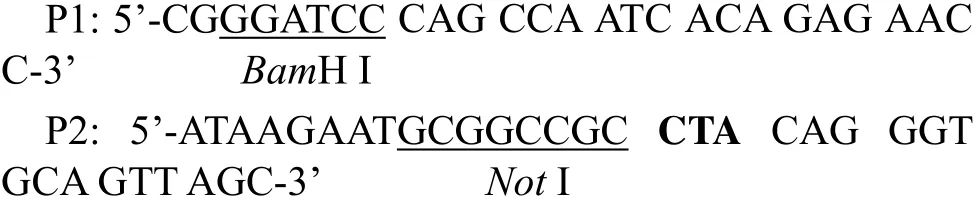
The total RNA was reversely transcribed using MMLV Single Step RT-PCR Kit (BBI,Canada).The fGH cDNA was modified by introducing aBamH I site at the 5’ end,and aNotI site and a TAG stop codon at the 3’ end.The amplified DNA bands were excised from the agarose gel and purified using AxyPrep DNA Gel Extraction Kit(Axygen,USA).The purified fGH cDNA was cloned into pMD18-T vector (Takara,Dalian,China) and then transformed intoE.coliDH5α.Five positive clones were selected randomly and sequenced using ABI 3730 sequencer.
2.3 Plasmid and Strains
Fusion expression plasmid pGEX-fGH was constructed by inserting the fGH cDNA into plasmid pGEX-4T-3,and then transformed intoE.coliDH5a.The positive clones(selected by PCR) were sent to the Invitrogen Company(Shanghai,China) for sequencing.BLAST was used to align the fGH cDNA sequence to those in NCBI in the Internet.
ThenE.coliBL21 (DE3) was transformed by the constructed plasmid and then used for expression of the fusion protein GST-fGH (Zanget al.,2005).
2.4 Expression of Fusion Protein
E.coliBL21 (DE3) containing pGEX-fGH plasmid was grown overnight at 37℃ in 2 ´ YTA medium containing 100 mg mL−1ampicillin.Cultures were then diluted (1:20) in fresh 2 ´ YTA medium and grown at 37℃for approximately 2 h until OD600reached approximately 0.5.The expression of r-fGH was initiated by the appending of isopropyl-β-D-thiogalactoside (IPTG) (Sigma,USA) to a final concentration of 0.1–2 mmol L−1.After 1–5 h induction,the cells were collected,disrupted by ultrasonication,and centrifuged at 12000×gfor 20 min.The supernatant and cell pellet were collected separately and electrophoresed on 12% SDS polyacrylamide gel electrophoresis (SDS-PAGE).The expressed protein was quantified using Quantity One (Bio-Rad,USA).The uninducedE.coliBL21 (DE3) / pGEX-fGH and the untransformedE.coliBL21 (DE3) served as negative controls.
2.5 Solubilization and Purification of Fusion Proteins from Inclusion Body
2.5.1 Collection of inclusion body of GST-fGH fusion protein
Two liters of cultured cells induced as described above were harvested by centrifugation at 10000×gfor 5 min.The pellet was resuspended in 200 mL of lysis buffer (pH 8.0,20 mmol L−1Tris,1 mmol L−1EDTA and 100 mg L−1lysozyme) and agitated at room temperature for 1 h.Thereafter,the inclusion body was precipitated and collected by centrifugation at 12000×gat 4 ℃ for 20min.
2.5.2 Denaturation of inclusion body
The inclusion body was washed twice with buffer I (20 mmol L−1Tris-HCl,1 mmol L−1EDTA,0.1 mol L−1NaCl,pH 8.0),twice with buffer Ⅱ (20 mmol L−1Tris-HCl,1 mmol L−1EDTA,0.1 mol L−1NaCl,2 mol L−1urea,pH 8.0),and once with buffer I,then solubilized by denaturing agent (8 mol L−1urea,20 mol L−1Tris-HCl,1 mmol L−1EDTA,0.5 mol L−1NaCl,0.1 mmol L−1DTT,pH 8.0) at room temperature for 1–2 h.The solution was centrifuged at 12000×gfor 10 min and the supernatant was collected.
2.5.3 Renaturatin of GST-fGH fusion protein by dilution
To renature the fusion protein,three steps were involved: (1) Addition of denatured GST-fGH fusion protein (2 mg mL−1) into renaturing buffer (20 mmol L−1Tris-HCl,1 mmol L−1EDTA,0.1 mol L−1NaCl,1 mmol L−1GSH,0.1 mmol L−1GSSG,pH 8.5) at the ratio of 1:80; (2) Addition of renaturing buffer (20 mmol L−1Tris-HCl,1 mmol L−1EDTA,0.1 mol L−1NaCl,1 mmol L−1GSH,0.1 mmol L−1GSSG,pH 8.5) into denatured GST-fGH fusion protein (2 mg mL−1); and (3) Additon of renaturing buffer (20 mmol L−1Tris-HCl,1 mmol L−1EDTA,0.1 mol L−1NaCl,1 mmol L−1GSH,0.1 mmol L−1GSSG,pH 7.0) into denatured GST-fGH fusion protein (2 mg mL−1).All the renatured solutions were gently agitated at 4℃ overnight.
2.5.4 Renaturatin of GST-fGH fusion protein by dialysis
Denatured GST-fGH fusion protein (1 mg mL−1) was dialyzed against 2 mol L−1urea renaturing buffer (20 mmol L−1Tris-HCl,1 mmol L−1EDTA,1 mmol L−1GSH,0.1 mmol L−1GSSG,2 mol L−1urea,pH 8.5),1 mol L−1urea renaturing buffer (20 mmol L−1Tris-HCl,1 mmol L−1EDTA,1 mmol L−1GSH,0.1 mmol L−1GSSG,1 mol L−1urea,pH 8.5),0.1 mol L−1urea renaturing buffer (20 mmol L−1Tris-HCl,1 mmol L−1EDTA,1 mmol L−1GSH,0.1 mmol L−1GSSG,0.1 mol L−1urea,pH 8.5),1 mmol L−1urea renaturing buffer (20 mmol L−1Tris-HCl,1 mmol L−1EDTA,1 mmol L−1GSH,0.1 mmol L−1GSSG,1 mmol L−1urea,pH 8.5),and PBS (pH 7.3) consecutively at 4℃ for 24 h.
2.5.5 Purification of GST-fGH fusion protein on Sepharase column
Renatured protein was incubated with Glutathione Sepharase 4B for 30 min and then loaded into column.After the column was washed twice with PBS (pH 7.3),the purified GST-fGH protein was eluted by elusion buffer at 20℃.Another manipulation method was the cleavage of the fusion protein by thrombin in column at 22℃ for 16 h.The collection from the column was used as the purified recombinant protein-fGH.
2.6 Purification of GST-fGH Fusion Protein from Soluble Extraction
One liter of cultured cells were induced by IPTG (0.2 mmol L−1) at 30℃ for 2 h and then harvested by centrifugation at 10000×gfor 5 min.The pellet was resuspended in 50 mL PBS (pH 7.3) and treated by ultrasonication.After centrifugation at 12000×gfor 20 min,the supernatant was collected and incubated with Glutathione Sepharase 4B for 30 min.The purified GST-fGH protein was eluted by elusion buffer at 20℃ and the purified r-fGH was cleaved by thrombin at 22℃.
2.7 Protein Content Calculation
Standard solution of albumin bovine was prepared and diluted at grads.OD280measurements were used for preparing the standard curve.The relationship between protein content and OD280conformed with 1 OD280(approximately 0.5 mg mL−1protein).

2.8 Western-Blotting Analysis of Protein Expression
The expressed proteins were transferred from the unstained SDS polyacrylamide gel to nitrocellulose sheets by electrophoresing.The filter was blocked overnight at 4℃ using a blocking solution.Immunoreactions were carried out with rabbit anti salmon GH polyclonal antibodies (1:1000,Gropep).Then,HRP-conjugated goat anti rabbit IgG secondary antibody was added.The filter was washed for four times,and the proteins were detected by color reaction with diaminobenzidine (DAB).
2.9 Receptor Binding Activity Analysis of Expressed r-fGH by ELISA-Receptor Assay
The receptor binding activity of r-fGH was tested by ELISA receptor assay (ELISA-RA) as described by Chenet al.(1995),herein with slight modifications.Liver of a liveP.olivaceuswas dissected and skived in liquid nitrogen,to which was added an extraction solution (0.02 mol L−1Tris-HCl,MgCl26 mmol L−1,saccharose 0.3 mol L−1)at a ratio of 1:10 (liver to buffer).The mixture was homogenized and centrifuged at 20000×gfor 20 min.The supernatant collected as coarse liver membrane receptor was diluted and coated onto 96-well ELISA plate at 4℃overnight.After blocking,the plate was used for ELISA-receptor assay.
Purified r-fGH proteins were added to the blocked plate and incubated at 37℃ for 2 h.Then,rabbit anti salmon GH polyclonal antibodies (1:1000) were added to the plate,followed by the addition of HRP-conjugated goat anti rabbit IgG secondary antibody.The plate was washed 3 times with PBST,and color reaction proceeded in darkness.
3 Results and Discussion
3.1 Construction of pGEX-fGH Expression Vector
Total RNA was isolated from the pituitary ofP.olivaceusand qualified for Reverse-Transcription PCR amplification (RT-PCR).The fGH cDNA of approximate 510 bp was amplified by RT-PCR and inserted into the multiple site of pGEX-4T-3 (Pharmacia,Sweden).The ligation mix was transformed into competent cells ofE.coliBL21(DE3) (Pharmacia,Sweden) and the recombinant clones were selected by PCR.After sequencing,BLAST search against the NCBI database indicated that the fragment was the fGH cDNA and the sequence was consistent with the sequence reported by Watahiki (1989).
3.2 Expression of Fusion Protein

Fig.1 SDS-PAGE analysis of protein expression.M,protein marker; 1,expression products of E.coli BL21 (DE3)/ pGEX-fGH without inducing; 2,expression products of E.coli BL21 (DE3) / pGEX-fGH induced by IPTG; 3,expression products of E.coli BL21 (DE3) / pGEX-4T-3 induced by IPTG; 4,whole cell lysates of E.coli BL21(DE3) / pGEX-fGH; 5,supernatant of ultrasonicated cells;and 6,pellet of ultrasonicated cells.
As shown in Fig.1,SDS-PAGE analysis of protein ex-tracted from the recombinantE.coliBL21 (DE3) / pGEX-fGH cells showed the presence of a specific 45 kD (GST 26 kD fused with r-fGH 19 kD) protein upon induction with IPTG.The 45 kD protein was tested mainly in precipitations,indicating that the fusion protein was primarily expressed as inclusion body.
3.3 Purification and Analysis of GST-fGH Fusion Proteins from Inclusion Body
After the lysis of the expressed cells and collection of the precipitations,inclusion body was obtained with a purity of 92% (Fig.2).Renaturation of inclusion body was performed in 8 mol L−1urea solution using the dilution and dialysis methods.The renatured protein was purified by Glutathione Sepharase 4B affinity chromatography.A single band of 45 kD protein was present on the SDSPAGE gel and the purity of the recombinant fusion protein was up to 97%.Cleavage of the fusion protein in chromatography column by thrombin yielded a specific 19 kD protein (Fig.2).Western-blotting analysis detected the 45 kD and 19 kD protein bands only on the film(Fig.2).The results showed that both the GST-fGH fusion protein and the recombinant fGH cleaved by thrombin specifically reacted with rabbit anti salmon GH polyclonal antibodies.The purified expression product from inclusion body was considered to be recombinant fGH protein.

Fig.2 SDS-PAGE and Western-blotting analysis of the purified r-fGH recovered from inclusion body.M,protein marker; 1,expression products of E.coli BL21 (DE3) /pGEX-fGH induced by IPTG; 2,inclusion body; 3,recovery product of GST-fGH; 4,purification product of r-fGH; 5,western-blotting analysis of purified GST-fGH;and 6,western-blotting analysis of purified r-fGH.
As shown in Table 1,the recovery rate of pH 8.5 renaturing buffer was higher than that of pH 7.0 renaturing buffer,whereas the dialysis method had the lowest recovery rate.It was obvious that the dilution method surpassed the dialysis method in renaturation of r-fGH.The fusion protein was likely unable to exist stably and tended to congregate under the effect of middle denaturing buffer.Using the dilution method,either addition of denaturing solution to renaturing buffer or addition of renaturing buffer to denaturing solution would require substantial time to allow the refolding of protein.The r-fGH may be more stable in relatively high pH environment(pH 8.5).The average yield of purified r-fGH from inclusion body was 2.605 mg L−1.
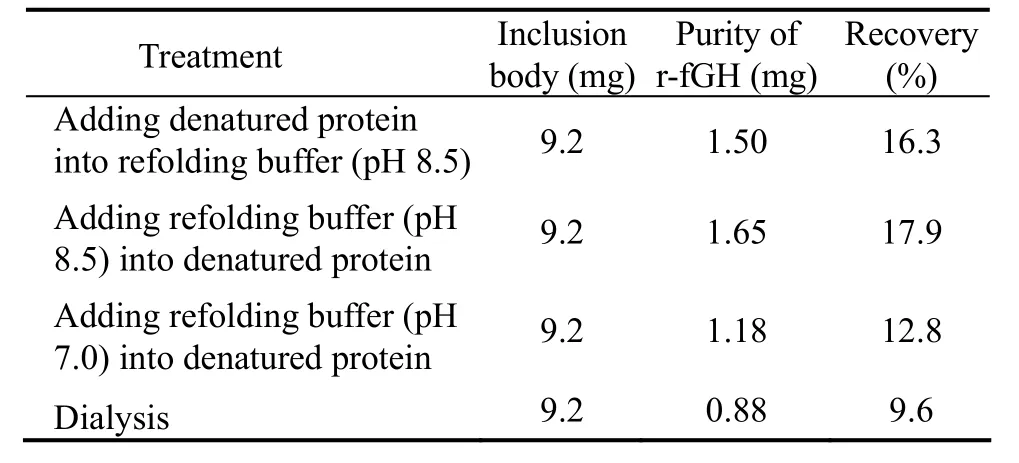
Table 1 Recovery rates of recombinant fGH from inclusion body using different treatments
3.4 Purification and Analysis of GST-fGH Fusion Proteins from the Soluble Product
According to our previous work (Zanget al.,2005),the expression of soluble r-fGH by pGEX-4T-3 can be improved by decreases in the inducing temperature and time,as well as the IPTG concentration.Under the conditions of 30℃ and 0.2 mmol L−1IPTG for less than 2 h,the percentage of soluble expression protein was up to 9.3%of the total protein.
For the soluble product,GST-fGH fusion protein can be directly purified from the lysates of inducedE.coliBL21 (DE3).After purification by an affinity column,the expression product was analyzed by SDS-PAGE and three bands (45 kD,26 kD and 19 kD) appeared (Fig.3).Western-blotting analysis showed that the purified product reacted with rabbit anti salmon GH polyclonal antibodies and two specific bands appeared at 45 kD and 19 kD,whereas the product cleaved by thrombin showed one band at 19 kD (Fig.3).These proved that the purified protein from the soluble expression product was recombinant fGH.
SDS-PAGE analysis of the purified protein from soluble product and inclusion body showed different bands,that is,three bands (45 kD,26 kD and 19 kD) from the former and only one band (45 kD) from the latter.It was likely that the soluble protein was unstable.GST-fGH (45 kD),GST (26 kD) and r-fGH (19 kD) composed of the three bands of soluble product detected by SDS-PAGE and the bands of GST-fGH (45 kD) and r-fGH (19 kD)were detected by western-blotting.The protein folded tightly in inclusion body,thus could not be easily degraded by proteinase.Consequently,SDS-PAGE and Western-blotting showed only one band of GST-fGH (45 kD).Although the products purified from inclusion body and soluble protein were not exactly the same,r-fGH (19 kD) was the only protein product obtained after cleavage by thrombin.The yield of r-fGH purified from soluble product was 1.964 mg L−1.

Fig.3 SDS-PAGE and Western-blotting analysis of purified r-fGH from the soluble protein.M,protein marker; 1,whole cell lysates of E.coli BL21 (DE3) / pGEX-fGH; 2,Cytoplasma of E.coli BL21 (DE3) / pGEX-fGH after ultrasonication; 3,purification product of GST-fGH; 4,purification product of r-fGH; 5,western-blotting analysis of purified GST-fGH; and 6,western-blotting analysis of purified r-fGH.
3.5 Receptor Binding Activity of r-fGH
ELISA-RA assay showed that the OD492of r-fGH increased with a growing r-fGH content when the ELISA plate was coated with flounder liver membrane receptor at a specific dilution (e.g.,1:50).Without the receptor coating treatment of ELISA plate,the OD492values remained at the background level regardless of the r-fGH content (Fig.4).However,the flounder liver membrane receptor was found to react with the primary antibody(rabbit anti salmon GH polyclonal antibodies) even with no addition of r-fGH.An increase in the receptor dosage resulted in elevated OD492values.Three ratios of dilution(1:5,1:20 and 1:80) of the receptor were used to analyze the relationship of r-fGH content with OD492values under different receptor dosages.The results showed consistent relationships of r-fGH content between OD492with three different dilutions ratios.The corresponding curves overlapped upon removal of the OD492background of receptor binding to primary antibody (Fig.5).This indicated that the bioactivity of r-fGH was unrelated with the concentration of receptor,and that it could be tested by ELISARA.
ELISA-RA showed that all the tested purification methods yielded r-fGH with biological activities,despite the fact that associated activities were unequal.Of these,the protein purified from soluble product had the best receptor binding activity,followed by that recovered by addition of renaturing buffer (pH 8.5) into the denatured product and the dialysis method.The products recovered by addition of denatured inclusion body into pH 7.0 or pH 8.5 renaturing buffer had the lowest receptor binding activity (Fig.6).Previous research has shown that addition of denatured product into renaturing buffer is an effective method for r-fGH purification.However,the activity of r-fGH recovered through this method was not good in the current study,even though the procedure was quite slow to prevent the wrong folding of the target protein.Of all the tested purification methods,the renaturing buffer of pH 8.5 was more advantageous than that of pH 7.0.It was likely that the isoelectric point of r-fGH was close to pH 7.0,allowing the protein to be easily congregating in solutions of pH at the isoelectric point (Marshaket al.,1996).By comparison,the recovery rate of fGH at pH 7.0 was lower and the associated receptor binding activity was weaker.

Fig.4 The binding curve of purified r-fGH to liver membrane receptor.◆: ELISA plate coated with liver membrane receptor; ▲: ELISA plate coated with skimmed milk.

Fig.5 The binding curve of r-fGH to liver membrane receptor of different concentrations.◆: 1:5 dilution ratio of receptor ; ▲: 1:20 dilution ratio of receptor ; ■: 1:80 dilution ratio of receptor.
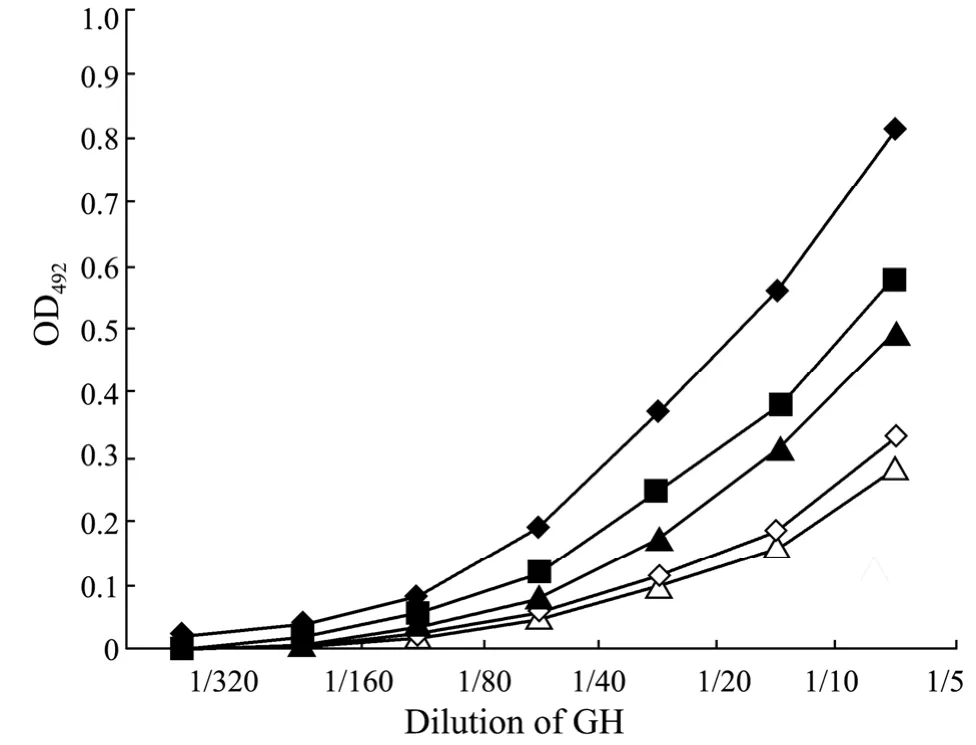
Fig.6 The binding curve of r-fGH purified by different treatments.◆,purified r-fGH from soluble product; ▲,purified r-fGH from the dialysis method; ■,purified r-fGH from the dilution method (adding pH 8.5 renaturing buffer into denatured solution); △,purified r-fGH from the dilution method (adding denatured solution into pH 8.5 renaturing buffer); ◇,purified r-fGH from the dilution method (adding pH 7.0 renaturing buffer into denatured solution).
4 Conclusions
This study demonstrated that the expression of fGH in pGEX-4T-3 was an efficient method for obtainment of r-fGH,and that the target protein could be expressed as inclusion body or in a soluble form.Comparison of the r-fGH yields and receptor binding activities of different purification methods indicated that when the product was mainly expressed as inclusion body,its recovery should be carried out by adding renaturing buffer (20 mmol L−1Tris-HCl,1 mmol L−1EDTA,0.1 mol L−1NaCl,1 mmol L−1GSH,0.1 mmol L−1GSSG) into the denatured solution.This yielded a recovery rate of up to 17.9%,and the purified protein had high bioactivity.The content of expressed soluble protein was increased by reducing the inducing time and temperature,as well as IPTG concentration.Despite the instability of soluble fusion protein,its bioactivity was higher than that purified from the inclusion body.This study proposed an optimized method for purification of r-fGH,which has the prospect of application to fish physiological study and flounder farming.
Acknowledgements
The work was supported by the National Natural Science Foundation of China (No.30901111),the China Agriculture Research System (CARS-50),and the key Project of Chinese Ministry of Education (No.108083).
Acosta,J.,Morales,R.,Morales,A.,Alonso,M.,and Estrada,M.P.,2007.Pichia pastorisexpressing recombinant tilapia growth hormone accelerates the growth of tilapia.Biotechnology Letters,29: 1671-1676.
Chan,Y.H.,Cheng,C.H.K.,and Chan,K.M.,2003.Recombinant goldfish growth hormones (gfGH-1 and -II) expressed inEscherichia colihave similar biological activities.Comparative Biochemistry and Physiology Part A,135: 613-624.
Chen,S.L.,Deng,W.T.,He,L.,and Chen,X.H.,1995.ELISA-receptor assay for testing the bioactivity of fish growth hormone.Journal of Fisheries of China,3: 217-224.
Jeh,H.S.,Kim,C.H.,Lee,H.K.,and Han,K.,1998.Recombinant flounder growth hormone fromEscherichia coli:overexpression,efficient recovery,and growth – promoting effect on juvenile flounder by oral administration.Journal of Biotechnology,60: 183-193.
Knobil,E.,and Hotchkiss,J.,1964.Growth hormone.Annual Review of Physiology,26: 47-74.
Li,Y.H.,Bai,J.J.,Jian,Q.,Ye,X.,Lao,H.H.,Li,X.H.,Luo,J.R.,and Liang,X.F.,2003.Expression of common carp growth hormone in the yeastPichia pastorisand growth stimulation of juvenile tilapia (Oreochromis niloticus).Aquaculture,216: 329-341.
Liu,S.M.,Zang,X.N.,Liu,B.,Zhang,X.C.,Arunakumara,K.K.I.U.,Zhang,X.Q.,and Liang,B.,2007.Effect of growth hormone transgenicSynechocystison growth,feed efficiency,muscle composition,haematology and histology of turbot(Scophthalmus maximusL.)Aquaculture Research,38: 1283-1292.
Marshak,D.R.,Kadonaga,J.T.,and Burgess,R.R.,1996.Production.In:Strategies for Protein Purification and Characterization: A Laboratory Course Manual.Cold Spring Laboratory Press,New York,396pp.
Mukhopadhyay,A.,1997.Inclusion bodies and purification of proteins in biologically active forms.Advances in Biochemical Engineering and Biotechnology,56: 61-109.
Paduel,A.,Chapnik-Cohen,N.,Gertler,A.,and Elizur,A.,1999.Preparation and characterization of recombinant dolphin fish (Coryphaena hippurus) growth hormone.Protein Expression and Purification,16 (3): 417-423.
Sekine,S.,Mizukami,T.,Nishi,T.,Kuwana,Y.,Saito,A.,Sato,M.,Itoh,S.,and Kawauchi,H.,1985.Cloning and expression of cDNA for salmon growth hormone inEscherichia coli.Proceedings of the National Academy of Sciences of the United States of America,82: 4306-4310.
Venugopal,T.,Anathy,V.,Pandian,T.J.,Gong,G.Z.,and Mathavan,S.,2002.Molecular cloning of growth hormoneencoding cDNA of India major carp,Labeo rohita,and its expression inEscherichia coliand Zebrafish.General and Comparative Endocrinology,125: 236-247.
Watahiki,M.,Ohara,E.,Tsuda,M.,Shoji,K.,Masuji,A.,Tanaka,M.,Yamakawa,M.,Ushiro,H.,Yoneda,Y.,and Nakashima,K.,1992.Syntheses of recombinant yellowtail and flounder growth hormones inEscherichia coli.Bioscience Biotechnology and Biochemistry,56: 1012-1016.
Watahiki,M.,Yamamoto,M.,Yamakawa,M.,Tanaka,M.,and Nakashima,K.,1989.Conserved and unique amino acid residues in the domains of the growth hormones.Journal of Biological Chemistry,264 (1): 312-316.
Zang,X.N.,Liu,B.,Zhang,X.C.,Mao,Y.X.,and Sui,Z.H.,2005.Cloning of cDNA ofParalichthys olivaceusgrowth hormone and its fusion expression inEscherichia coli.High Technology Letters,3: 99-104.
 Journal of Ocean University of China2013年1期
Journal of Ocean University of China2013年1期
- Journal of Ocean University of China的其它文章
- Isolation and Expression Analysis of FTZ-F1 Encoding Gene of Black Rock Fish (Sebastes schlegelii)
- Chemical Characteristics and Anticoagulant Activities of Two Sulfated Polysaccharides from Enteromorpha linza(Chlorophyta)
- Seasonal Changes in Food Uptake by the Sea Cucumber Apostichopus japonicus in a Farm Pond: Evidence from C and N Stable Isotopes
- Purification and Characterization of a New Thermostable κ-Carrageenase from the Marine Bacterium Pseudoalteromonas sp. QY203
- Growth,Metabolism and Physiological Response of the Sea Cucumber,Apostichopus japonicus Selenka During Periods of Inactivity
- What Depth Should Deep-Sea Water be Pumped up from in the South China Sea for Medicinal Research?