
醋酸杆菌Acetobacter xylinum pde基因敲除研究
2013-01-29 05:59:28齐香君李彦军
陕西科技大学学报 2013年5期
张 雯, 齐香君, 李彦军
(陕西科技大学 生命科学与工程学院, 陕西 西安 710021)
0 引言
细菌纤维素(Bacterial cellulose, 简称BC)是由醋酸杆菌(Acetobacterxylinum)高效合成的纤维素,其最终合成依赖于纤维素合成酶完成,纤维素合成酶表达量的多少,酶活力的高低,直接影响到BC的合成及产量高低.环鸟苷酸(cGMP)是纤维素合成酶的变构激活剂,如果纤维素合成酶的变构位点上没有结合cGMP,纤维素合成酶将不具有活性.cGMP的合成与降解又与鸟苷酸环化酶(GC)和磷酸二酯酶(PDE)有关.GC催化cGMP的合成, PDE催化cGMP的降解[1-4].
欲提高纤维素合成酶活性,最有效的途径就是消除PDE对cGMP的降解作用,增加其胞内含量,提高细菌纤维素合成酶活力,最终提高BC产量.本研究通过将氨苄抗性基因(Ampr基因)插入至PDE编码基因(pde基因)BanⅡ、BssHⅡ酶切位点(所得重组基因记为pde′),使pde基因失活,将重组基因pde′克隆至质粒pHSG398 EcoRⅠ、KpnⅠ酶切位点,构建重组质粒pde′-pHSG398.pde′-pHSG398转化醋酸杆菌,利用同源双交换使重组细胞内pde′替换染色体上的pde基因,从而敲除醋酸杆菌的pde基因,使其丧失表达PDE的功能,构建PDE失活型(PDE-)重组菌株,并利用加入氨苄青霉素及氯霉素的培养基筛选出PDE-重组菌株.
1 材料与方法
1.1 材料与仪器
醋酸杆菌(Acetobacterxylinum) 由本实验室分离纯化而得;质粒pHSG398 日本Takara生物公司;限制性核酸内切酶BanⅡ、BssHⅡ、KpnⅠ、EcoRⅠ、Taq酶、T4连接酶、dNTPs、DNA Marker DL2000、Wide Range DNA Marker宝生物工程(大连)有限公司;Ampr基因 由质粒pUC19经PCR扩增得到;pde基因引物、Ampr基因引物 利用primmer5.0软件设计,由宝生物工程(大连)有限公司合成;氨苄青霉素、氯霉素.山东鲁抗医药集团有限公司;固体培养基:蔗糖5%,牛肉膏1.5%,Na2HPO40.44%,柠檬酸0.08%,琼脂1.8%,无水乙醇1.0%(v/v),pH自然;发酵培养基:蔗糖5%,牛肉膏1.5%,Na2HPO40.44%,柠檬酸0.08%,无水乙醇1.0%(v/v),pH自然;氨苄青霉素培养基:蔗糖5%,牛肉膏1.5%,Na2HPO40.44%,柠檬酸0.08%,琼脂1.8%,氨苄青霉素0.05%,无水乙醇1%(v/v);氯霉素培养基:蔗糖5%,牛肉膏1.5%,Na2HPO40.44%,柠檬酸0.08%,琼脂1.8%,氯霉素0.05%,无水乙醇1%(v/v).
PCR仪(MJ PTC-200) 美国MJ Research公司;电泳仪(DYY-10C)、电泳槽(DYCP-31D) 北京市六一仪器厂;高速台式冷冻离心机(TGL-16M) 长沙湘仪离心机仪器有限公司;高速离心机(TGL-16C) 上海安亭科学仪器厂;电热恒温培养箱(MG250B)、恒温振荡器(HYG-1A) 上海新瑞仪器有限公司;光学显微镜(XPS-8CA) 上海光学仪器有限公司;傅立叶变换红外光谱仪 德国Brucher公司.
1.2 分析方法
(1)DNA检测[5]:琼脂糖凝胶电泳法(电泳参数:U=60 V,I=50 mA,P=50 W);
(2)BC膜处理[5]:将发酵所得BC膜浸泡于0.1 mol/L NaOH溶液中,80 ℃浸泡30 min,继续加热煮沸约2 h,再用蒸馏水反复冲洗,直到凝胶膜透明无色,pH为7.2;
(3)BC产量的测定[5]:静态发酵BC膜处理后,用干燥滤纸吸干表面水分,105 ℃干燥至恒重,称重.单位:g/L培养液;
(4)BC鉴定及基团分析[6,7]:红外光谱(样品处理:将处理好的BC膜烘干后研成粉末,与溴化钾以1∶100比例混合,充分研细,然后用压片机压片,再放入红外光谱仪中进行测定).
1.3 实验方法
1.3.1 菌体细胞的收集及染色体DNA的提取[8,9]
取一定量醋酸杆菌种子培养液涂布于固体培养基上,30 ℃培养24 h,刮菌膜于无菌水中,漩涡混合振荡,脱脂棉过滤,12 000 rpm离心10 min收集沉淀,利用酚-氯仿抽提法提取染色体DNA.
1.3.2 醋酸杆菌pde基因的敲除[9,10]
(1)pde′基因的构建及克隆
根据pde基因序列及Ampr基因序列[11]设计PCR引物,如表1所示.为便于pde′基因的构建及与质粒pHSG398的克隆,在Ampr基因PCR引物两端分别添加限制性核酸内切酶BanⅡ、BssHⅡ识别序列,在pde基因引物两端分别添加限制性核酸内切酶EcoRⅠ、KpnⅠ识别序列.PCR反应过程中分别设置Mg2+浓度1 mM、2 mM、3 mM、4 mM,退火温度46 ℃、48 ℃、50 ℃、52 ℃、54 ℃,考察其对PCR产物产量及特异性的影响.采用BanⅡ、BssHⅡ双酶切分别切割pde基因、Ampr基因,利用T4连接酶将Ampr基因连接至pde基因BanⅡ、BssHⅡ酶切位点(所得重组基因记为pde′).采用EcoRⅠ、KpnⅠ双酶切分别切割pde′基因、质粒pHSG398,将pde′基因连接至pHSG398EcoRⅠ、KpnⅠ酶切位点,构建重组质粒pde′-pHSG398.

表1 扩增基因PCR引物序列
注:(1)引物的酶切位点用下划线标识
(2)PDE-重组细胞的构建及筛选
采用CaCl2转化法使重组质粒pde′-pHSG398转化醋酸杆菌细胞,利用pde′基因上所带的氨苄抗性标记及质粒pHSG398上的氯霉素抗性标记,采用影印法将转化液分别涂布于氨苄青霉素培养基及氯霉素培养基,30 ℃静置培养24 h,筛选在氨苄青霉素培养基上生长而在氯霉素培养基上不生长的菌落30株,分别记为C1~C30,即为目的转化子,即PDE-重组细胞.
1.3.3 PDE-重组菌株的发酵[5]
配制发酵培养基,分别接种醋酸杆菌出发菌株(Q)及PDE-重组菌株(C1~C30),(接种量10%,发酵液装量200 mL/500 mL烧杯),30 ℃静态培养4 d进行BC的发酵生产(每个菌株3个平行实验),发酵结束后将BC凝胶膜按1.2.3所述方法进行处理,测定BC产量并进行BC膜IR检测.
1.3.4 PDE-重组菌株的遗传稳定性研究
将PDE-重组菌株(C1~C30)分别接种于氨苄青霉素培养基及氯霉素培养基上,连续转接三代,验证重组菌株的遗传稳定性能.
2 结果与讨论
2.1 Mg2+浓度、退火温度对pde基因、Ampr基因PCR扩增的影响
2.1.1 Mg2+浓度对PCR的影响
按照1.3.2.1所述方法考察Mg2+浓度对pde基因、Ampr基因PCR扩增的影响,实验结果如表2所示.PCR产物结果表明,随着Mg2+浓度升高,PCR产物产量逐渐增多,而产物特异性逐渐降低,在Mg2+较高时,甚至出现多条非特异性杂带.其原因为Taq酶活性的强弱依赖于游离Mg2+浓度的高低,从而影响PCR产物的产量和质量,而PCR反应体系中,模板的纯度、dNTPs的含量以及引物浓度均会影响游离Mg2+浓度.实验结果表明,pde基因PCR反应体系中Mg2+最佳浓度为3 mM,Ampr基因PCR反应体系中Mg2+最佳浓度为2 mM.
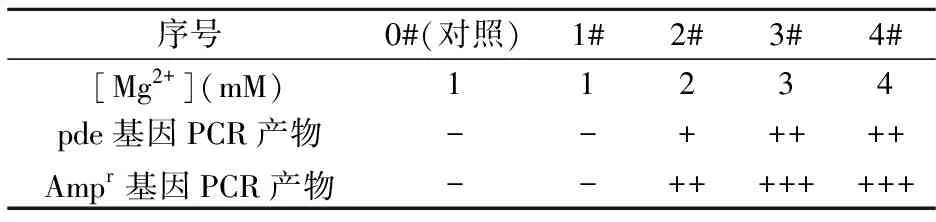
表2 Mg2+浓度对pde基因、Ampr基因PCR产物的影响
-:产物量极少;+:产物量较大,特异性好;++/+++:产物量大,但非特异性产物较多
2.1.2 退火温度对PCR的影响
按照1.3.2.1所述方法考察退火温度对pde基因、Ampr基因PCR扩增的影响,实验结果如表3、表4所示.PCR产物结果表明,pde基因PCR反应退火温度为50 ℃时,PCR产物产量相对较高,且特异性好;Ampr基因PCR反应退火温度为36 ℃时,PCR产物产量相对较高,且特异性好.其原因为在PCR反应中,退火温度的高低决定着PCR产物的产量及特异性,复性温度过低,引物之间易形成引物二聚体,或引物与模板易发生非特异性结合,形成非特异性产物;温度过高,引物与模板结合效率降低,PCR产物产量降低.

表3 不同复性温度对pde基因PCR产物的影响
注:-:产物量极少或没有;+:产物量较大,特异性好;++/+++:产物量大,但非特异性产物较多

表4 不同复性温度对Ampr基因PCR产物的影响
注:-:产物量极少或没有;+:产物量较大,特异性好;++/+++:产物量大,但非特异性产物较多

图1 不同复性温度下pde基因PCR产物电泳图
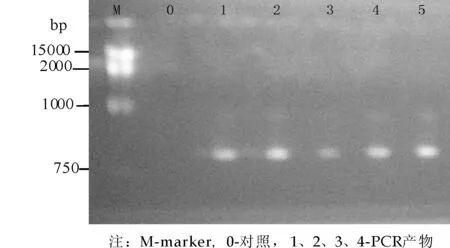
图2 不同复性温度下Ampr基因PCR产物电泳图
pde基因、Ampr基因PCR反应产物凝胶电泳图谱如图1、图2所示.由图可知,pde基因PCR产物大小介于500 bp~750 bp之间,符合pde基因671 bp的大小;Ampr基因PCR产物稍大于750 bp,符合Ampr基因856 bp的大小.
2.2 pde基因的敲除及克隆
根据1.3.2.1所述方法构建pde′基因.pde基因、Ampr基因酶切及连接前后电泳图谱,pde′基因、质粒pHSG398酶切前后电泳图谱,以及重组质粒pde′-pHSG398电泳图谱如图3、图4、图5所示.由电泳图谱可知,pde基因、Ampr基因进行了正确切割及连接,重组质粒pde′-pHSG398也进行了正确构建.
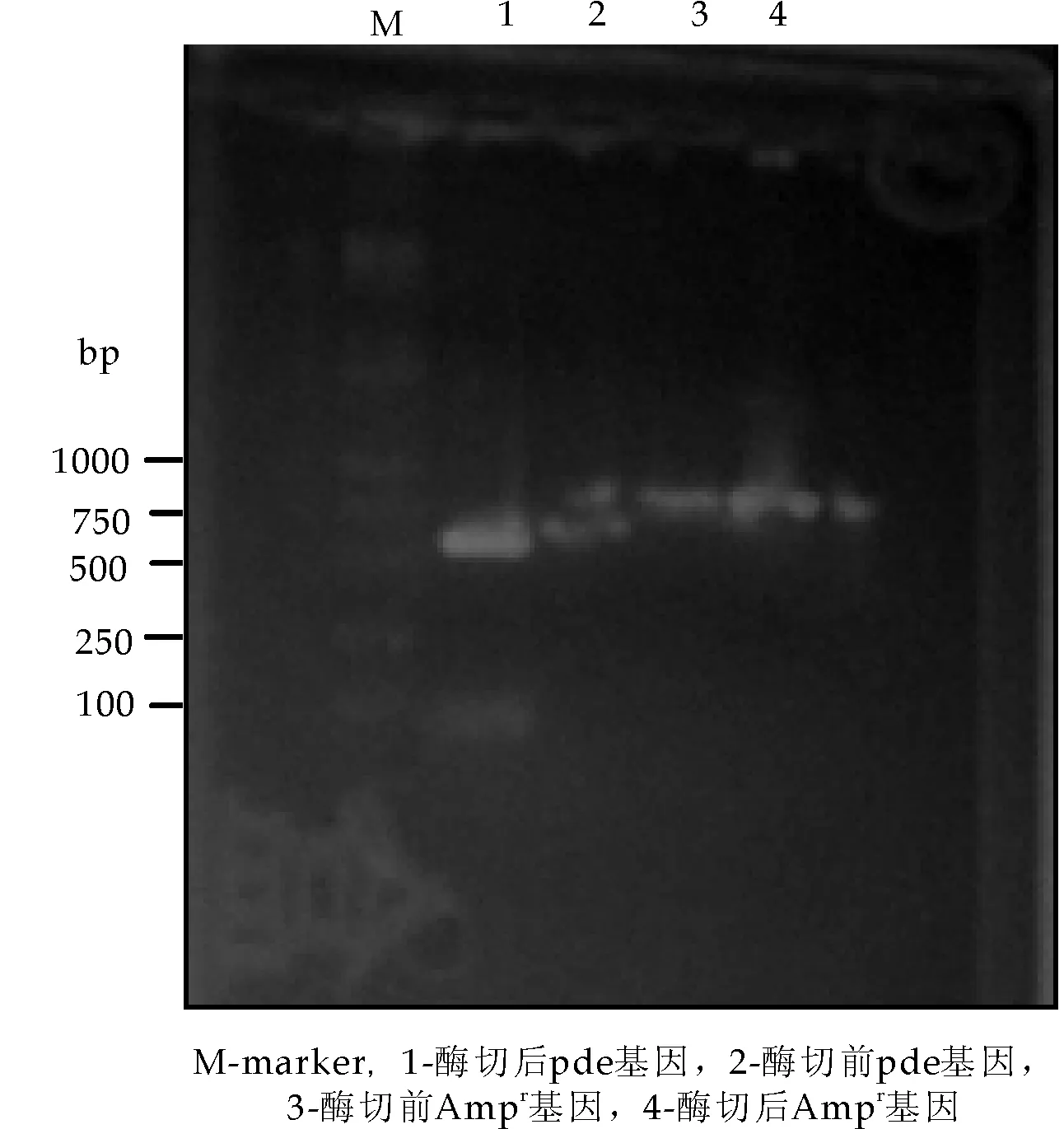
图3 pde基因、Ampr基因酶切前后电泳图谱

图4 pde基因、Ampr基因重组前后电泳图谱

图5 pde′基因、pHSG398重组前后电泳图谱
2.3 PDE-重组细胞的构建及筛选
根据1.3.2.2所述方法,采用CaCl2转化法,构建PDE-重组细胞.利用氨苄青霉素培养基及氯霉素培养基筛选目的重组细胞,共筛选得到30株PDE-重组细胞.
由于DNA发生同源重组时,同时发生双交换和单交换,仅发生单交换时,重组细胞基因组中不仅含有pde′基因,还含有宿主细胞的pde基因,因此需筛选发生双交换的转化子.双交换的转化子具有Ampr和Clms,因此筛选指标为在加有氨苄抗生素的培养基上生长而在加有氯霉素的培养基上不生长的菌落,即为目的重组菌株.
2.4 PDE-重组菌株的发酵
醋酸杆菌出发菌株(Q)及PDE-重组菌株(C1~C30)发酵生产BC产量如表5所示.出发菌株(Q)及PDE-重组菌株(C9)发酵产物BC红外图谱如图6、图7所示.由表5可知,所筛选30株PDE-重组菌株发酵产BC与醋酸杆菌出发菌株相比较,产量均有所提高,最大产率提高了38%,表明pde基因的敲除可提高BC合成酶作用水平,从而提高BC产量.由图6、图7可知,PDE-重组菌株(C9)发酵产物BC膜红外光谱图与出发菌株(Q) 发酵产物BC膜红外光谱图特征吸收峰相似.1 059 cm-1左右处的吸收峰是由碳氧键的伸缩振动引起的,是纤维素分子的特征峰.在3 450 cm-1左右处的吸收峰,反映了分子间氢键引起的O-H基的伸缩振动.在1 500 cm-1和2 000 cm-1之间的吸收峰是由纤维素4′端的半缩醛基引起的.在1 645 cm-1左右处的吸收峰是由C-H键伸缩振动引起的.红外光谱图显示pde基因敲除不影响菌株的BC合成能力.PDE-重组菌株(C1~C30)遗传稳定性实验结果显示,重组菌株接种于氨苄青霉素培养基及氯霉素培养基上,连续转接三代,仍然具有Ampr和Clms,证明其pde-基因型稳定,重组菌株遗传稳定性能良好.

表5 PDE-重组菌株与出发菌株BC产量的比较

图6 出发菌株发酵BC红外光谱图
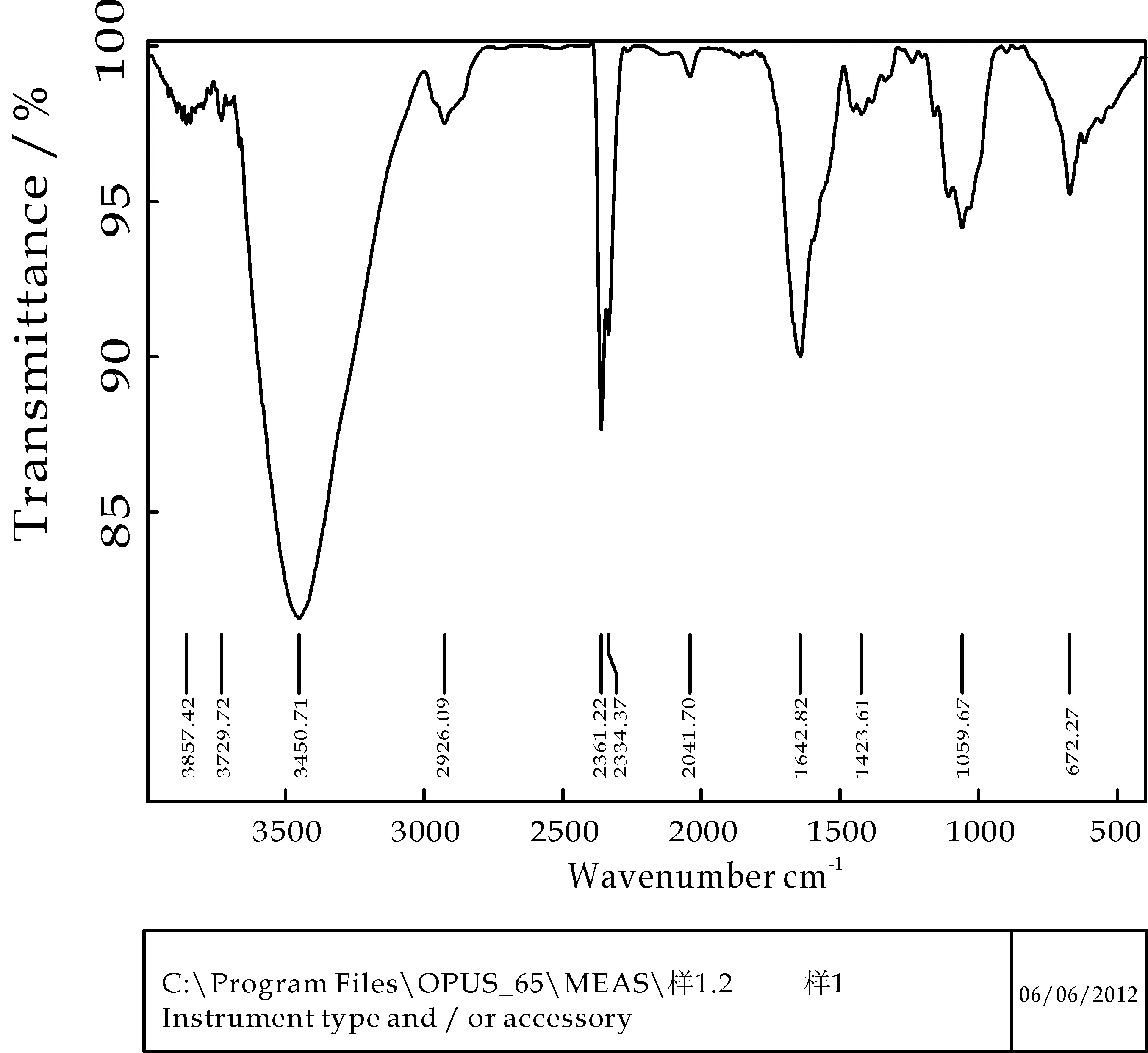
图7 PDE-重组菌株(C9)发酵BC红外光谱图
3 结束语
通过基因敲除技术能够敲除醋酸杆菌(Acetobacterxylinum)基因组中pde基因,成功构建PDE-菌株,重组菌株具备BC高产能力,且遗传稳定性能良好.本研究为改善细菌纤维素发酵产量低、细菌纤维素生产难以工业化的现状奠定了基础.
[1] Satoshi Kimura,Tetsuo Kondo.Recent progress in cellulose biosynthesis[J]. J Plant Res,2002,115(6):297-302.
[2] Neta Holland,Doron Holland,Tim Helentjaris.A comparative analysis of the plant cellulose synthase (CesA) gene family[J].Plant Physiology,2000,123(2):120-134.
[3] 齐香君,张 雯.功能性食品-纳塔的生物合成及生产菌基因构造[J].食品研究与开发,2004(S1):57-59.
[4] Bungay HR,Serafica.Methord for preparation of microbial cellulose[J].US Patent,2000,173(6):717-727.
[5] 张 雯,齐香君,韩戌珺.细菌纤维素生产菌株的动力学研究[J].食品科学,2005,26(12):65-67.
[6] 潘 颖.细菌纤维素的制备及改性研究[D].青岛:青岛大学,2007.
[7] Chao Y,Ishida Tsugano Y,Shoda M.Bacterial cellulose production by Acetobacter xylinum in a 50-1 internal-loop airlift reactor[J].Biotechnology Bioeng,2000,68(10):345-352.
[8] 张 雯,齐香君.细菌纤维素生产菌株菌体细胞收集方法的研究[J].食品工业科技,2006,27(9):57-58.
[9] 张维铭.现代分子生物学实验手册[M].北京:科学出版社,2003.
[10] 陈三凤,刘德虎.现代微生物遗传学[M].北京:化学工业出版社,2003.
猜你喜欢
纺织科技进展(2021年3期)2021-06-09 08:07:14
陶瓷学报(2021年1期)2021-04-13 01:33:02
食品科学(2018年10期)2018-05-23 01:27:28
西南医科大学学报(2015年1期)2015-08-22 13:01:46
中国当代医药(2015年9期)2015-03-01 02:01:59
西南军医(2015年6期)2015-01-23 01:25:50
应用化工(2014年11期)2014-08-16 15:59:13
食品工业科技(2014年9期)2014-03-11 18:15:28
河南医学研究(2014年2期)2014-02-27 14:51:32
浙江中西医结合杂志(2014年8期)2014-01-22 16:41:21
