
多巴胺构象结构和性质的密度泛函理论研究
2013-01-21 03:17:12黄旭慧孙晓玲蔡跃飘王朝杰
温州医科大学学报 2013年1期
黄旭慧,孙晓玲,蔡跃飘,王朝杰
(温州医学院 药学院,浙江 温州 325035)
药物分子的药理活性与其分子构型或构象结构有着十分密切的关系[1-2]。研究药物分子电子结构以及药物分子各种构象性质,可定量地分析药物结构-活性关系,为药物分子与生物体相互作用提供理论依据[3]。
多巴胺(dopamine,DA),化学名为邻苯二酚乙胺,系统名称为4-(2-乙胺基)苯-1,2-二酚。它是一种中枢神经系统的重要神经递质,主要参与运动、情感和神经内分泌的调节[4],临床研究报道较多。曹磊等[5]观察到小剂量多巴胺联合前列地尔能有效延缓肾功能不全进展。杨丽娟等[6]发现硫酸镁联合多巴胺对新生儿低氧低血性脑病(HIE)患儿有脑保护作用,同时减少了单用硫酸镁的不良反应。多巴胺的检测和结构性质研究引起许多学者的兴趣[7-10]。吴莹等[11]用半微分循环伏安法(SCV)对多巴胺在酶催化下与二茂铁[Fe(C5H5)2]在水/硝基苯(W/NB)界面发生电子传递的行为进行了研究,得到了良好的半微分极谱峰。Solmajer等[12]在pH为2~11.5范围内研究了DA构象的平衡性,结果发现在高pH值时,反式异构体更稳定,然而在低pH值时其他构象优于反式异构体。李志锋等[13]应用密度泛函理论(DFT),自然键轨道理论(NBO)及分子中的原子理论(AIM),对DA的构象稳定性及其相互转换的势能面进行了报道。Urban等[14]用AM1-SM1模型[15]对中性及电离的DA分子进行了理论计算,表明了溶剂效应对构象稳定性有重要影响。
DA在不同溶剂中的构象变化及对应性质方面的理论研究报道甚少,本工作基于密度泛函理论在量子化学计算上的优点[16-17],采用杂化密度泛函方法B3LYP对DA的不同构象进行结构全优化,获得最稳定结构DA1,详细计算分析了DA1在不同溶剂中构象变化引起的性质变化状况。
1 计算方法
本研究运用B3LYP方法[18-20]在6-311++G(2 d,p)基组水平上对气相中DA系列初始结构进行全优化和振动分析,得到11种稳定结构,其中能量最低是DA1。以DA1中二面角φ(C4-C9-C10-N11)为变量,5°旋转为步长,详细考察了0°~360°范围内构象变化引起的DA1几何结构、电子结构、能量学及振动光谱,并用TD-B3LYP计算了紫外-可见光谱(UV-Vis)性质,采用OPBE/6-311++G(2 d,p)//B3LYP/6-311++G(2 d,p)计算二面角φ变化引起的1H、13C-NMR数据[21],对各φ点对应构象结构运用概念密度泛函理论(C-DFT)进行了反应性分析。不同溶剂将对DA1的构象变化产生不同影响,在相同计算水平上采用极化连续模型(PCM)计算了DA1构象异构体在油相环己烷和水相中的上述性质变化。
有关C-DFT简述如下,Parr等[22]定义了化学势μ,化学硬度η,电负性χ,亲电子反应指数ω,按照化学势和化学硬度,ω=。根据Mulliken[23]的近似假设,μ=-χ=-(I+A),η=I-A。按照Koopmans理论假设[24],I≈-EHOMO,A≈-ELUMO,从而计算上述各量。所有计算均在Dell精密工作站上用G03程序包[25-28]完成的。
2 结果和讨论
2.1 多巴胺构象计算
2.1.1 几何结构:图1绘出了B3LYP/6-311++G(2 d,p)水平上气相中DA的11种不同稳定结构,这11种结构可绕相应C1-O7[二面角φ1:H15-O7-C1-C6]、C2-O8[二面角φ2:H16-O8-C2-C3]和C9-C10[二面角φ:C4-C9-C10-N11]单键旋转而相互转化,二面角φ1,φ2和φ的旋转是引起构象变化的主要原因。按照相对能量高低及苯环上两个羟基H的空间取向依次编号为DA1~DA7、DA1'、DA3'~DA5',分子中原子标示如结构DA1,在图中标明了不同结构中的O-H和N-H键长,以及分子内氢键O…H-O、N…H-C有关数值,单位为(1=10-10m)。
从图1可见,DA1~DA6中均能形成O…H-O型分子内氢键,由于分子内氢键的存在,在构象中均能形成一个五元环,该环与苯环处在同一平面上,容易形成大的p-π-p共轭体系。在DA4和DA5中,由于N9上有孤对电子,构象中存在N…H-C型分子内氢键,DA4中形成一个扭曲的四元环,而DA5中形成了一个扭曲的六元环。虽然N…H-C型分子内氢键的存在,但同时N9的孤对电子(LP)与苯环的π电子对间的排斥作用增加,而DA6中虽无N…H-C型氢键的形成,但其两个亚甲基-CH2-上的H原子与相邻苯环上的H产生排斥作用,因而能量升高,稳定性更低,而DA7相对于DA1无分子内氢键。由于苯环上两个羟基H的空间朝向不同也会引起结构稳定性的改变,我们用同样的方法在相同基组上对DA1~DA7中H15相对于C1-C2为顺式,H16相对于C2-C3为顺式的所有结构进行了几何全优化,结果只得到了 4 种稳定结构(DA1'、3'、4'和 5')。
2.1.2 能量学分析:对于得到4种羟基氢朝向不同稳定结构(DA1'、3'、4'和5'),分析其电子能量和吉布斯自由能,结果表明苯环上两个羟基H的空间朝向对构象的能量影响不大,DA1'与DA1两者相对电子能量的差值为0.46 kJ·mol-1,相对吉布斯自由能的差值为0.94 kJ·mol-1,两者相差数值很小,因此下文讨论中将不考虑羟基H的空间朝向对构象转化的影响,只分析7种稳定结构即DA1~DA7。
表1列出了DA1~DA7的能量学相关数据,其中电子能量E是在考虑了同水平B3LYP/6-311++G(2 d,p)下的零点振动能(ZPVE)校正后的数值(熵S除外),相对能Δ E可由下式定义:
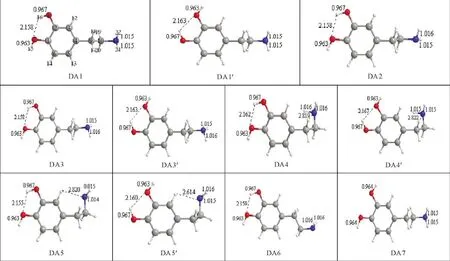
图1 B3LYP/6-311++G(2 d,p)水平上的DA稳定结构
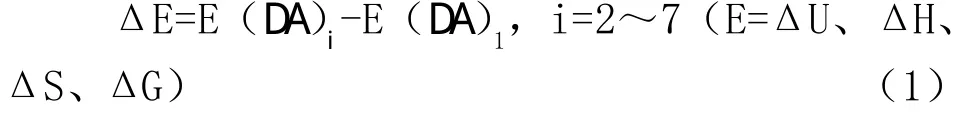
表1中的热力学函数相对值均可用上式计算。
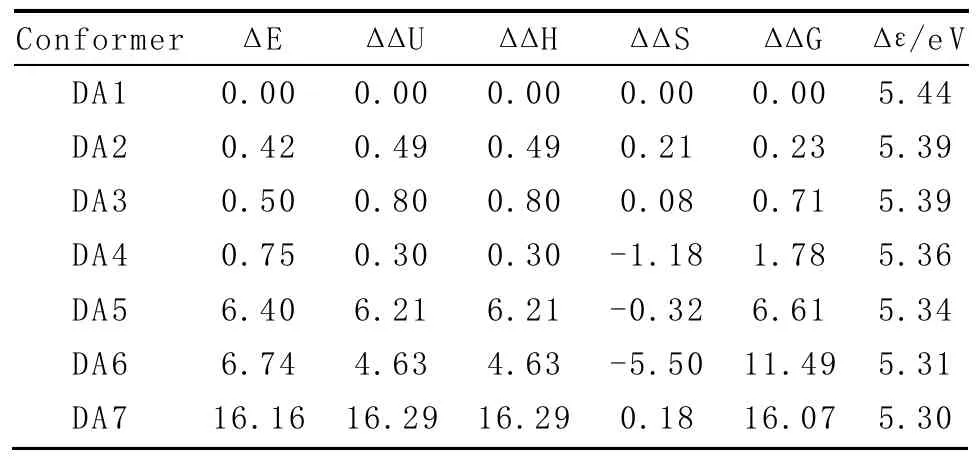
表1 B3LYP/6-311++G(2 d,p)计算DA1~DA7的相对热力学参数值(单位:kJ·mol-1)
表1给出了1atm和298.15 K下的气相中7种DA稳定结构的相对电子能量Δ E,相对内能Δ Δ U,相对焓Δ ΔH,相对熵Δ ΔS和相对吉布斯自由能Δ ΔG。DA1~DA4的相对能差各项值较小,稳定性相近。考虑了焓和熵的吉布斯自由能差Δ ΔG,则预示DA1在常温存在的概率最大。DA1与DA2和DA3相比,主要是由于DA1分子中-NH2空间排列,其中φ=180°达到了一种反式垂直的结构,结果电子能量降低,结构更加稳定,而DA2和DA3在结构上仅是-NH2在空间上的取向不同,其他结构参数都非常接近。DA4和DA5中二面角C4-C7-C8-N9仅相差10°,但能量差值却相差较大,主要是由于DA4中有π…H-N型氢键作用,而DA5中存在π…LP的排斥作用。DA6和DA7的能量差值都较大,尤其是DA7,与其他6种稳定结构相比,由于分子中没有形成分子内氢键,而其他结构参数与DA1也接近。同时表1列出了DA1~DA7的HOMO-LUMO前线分子轨道能级差Δ ε,Δ ε的数值变化范围在5.44~5.30 eV之间,随着不同结构的相对吉布斯自由能逐渐升高而呈相反变化,但彼此数值相近。
2.1.3 振动光谱:在对DA进行系列结构优化的基础上,用同样的方法和基组对DA1~DA7进行振动光谱和相对强度的计算,对七种稳定结构的特征峰进行分析,在3 483~3 491、1 465~1 560、1 657~1 662和3 027~3 050 cm-1范围内各有四个很强的伸缩振动峰,根据简正振动分析,它们分别对应O7-H15,苯环上C1-C2,C1-O7和C6-H14的伸缩振动。实验报道的数据[29]分别为νO7-H15=3368、νC1-C2=1498、νC1-O7=1614、νC6-H14=2 498 cm-1。利用文献报道的在该方法和基组下的振动频率校正因子为0.9692[30],对各计算值经校正后相对应的四个强峰波数范围是3 376~3 384、1 420~1 512、1 606~1 611和2 934~2 956 cm-1,各计算值与实验值的绝对误差在0~16 cm-1范围之内,与实验数值吻合很好。
2.2 DA1的构象变化和性质研究 首先在气相中,以二面角φ(C4-C9-C10-N11)为变量,每5°旋转为步长,在B3LYP/6-311++G(2 d,p)水平上计算0°~360°范围内DA1构象变化异构体的几何参数、能量学参数,用TD-B3LYP/6-311++G(2 d,p)计算UV-Vis光谱,基于文献[21]报道用OPBE/6-311++G(2 d,p)计算各构象异构体的1H和13C-NMR谱。其次运用PCM对DA1的各异构体在油相环己烷和水相中进行同样研究。下文将从溶剂效应对DA1构象变化的能量学、极性和反应性的影响,构象变化的UV-Vis光谱和NMR谱进行分析。
2.2.1 溶剂效应:图2以水相中最稳定构象即φ=180°的电子能量为基准,计算了DA1在气相(gas phase,g)、油相环己烷(cyclohexane phase,c)和水相(water phase,w)不同环境中DA1各种典型构象的相对电子能量Δ E ,并标出了0°~360°范围内曲线对应的部分极大值和极小值结构图。图上各驻点结构数据表明,构象变化随溶剂极性而改变,DA1不同构象中原子间的键长和键角都稍有变化。随着溶剂极性增大,相同驻点DA1中O-H、N-H和CN间的键长逐渐增大,∠HOC也随之增大。
图2中也标示了不同范围内DA1不同构象间在三相中的构象转化的能垒值E(g,w,c):当0°≤φ≤60°时,DA1构象转化的能垒随着介质的极性增加而增加,E1g=19.27<E1c=19.50<E1w=20.43 kJ·mol-1;而从60°<φ≤120°时,转动能垒则呈相反变化E2g=15.17>E2c=14.62>E2w=13.12 J·mol-1;当120°<φ≤180°时,E3g=15.67<E3c=16.07<E3w=16.22 kJ·mol-1。在气相中,DA1构象变化能阈为19.87 kJ·mol-1,在油相环己烷和水相中分别升高了1.08和3.66 kJ·mol-1。在0°~60°范围内,水的溶剂化效应使各个构象能量差距扩大,而在60°~120°范围内,则刚好相反。与气态条件相比,水相和油相环己烷的溶剂化效应对DA1的构象稳定性产生了不同程度的影响,较相应的气相具有更低的能量。同时图2表明,水和环己烷的溶剂化效应增加了DA1转换过程中的能垒,使构象转换在溶液中一般较真空中难。
为进一步分析0°~360°范围内,DA1在三相中不同构象间的相对电子能与φ之间的关系,用origin软件对其进行三角函数拟合,得到的拟合函数方程如下:

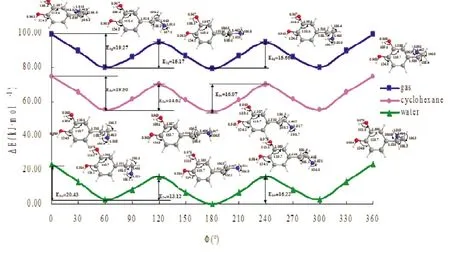
图2 B3LYP/6-311++G(2 d,p)水平上DA1在三相中的相对能量
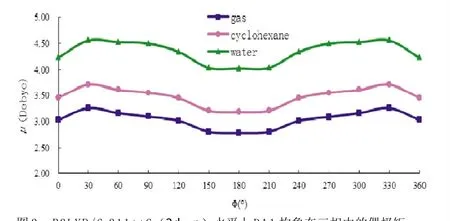
图3 B3LYP/6-311++G(2 d,p)水平上DA1构象在三相中的偶极矩
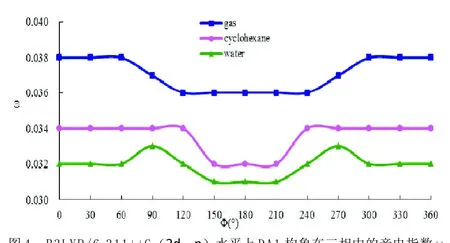
图4 B3LYP/6-311++G(2 d,p)水平上DA1构象在三相中的亲电指数ω
对DA1构象在三相中变化引起的偶极矩改变进行了计算分析,从图3可以看出,在油相环己烷及水相中,溶剂化效应通过诱导作用使不同构象的偶极矩增加,且极性较强的溶剂水对DA1的影响大于环己烷。由于DA1本身是极性分子,其气相真空中最稳定构象的μ=2.78D,在极性较小的油相环己烷介质中其偶极矩平均增加约0.43D,在极性强的水中增加达到1.30D,而水相与油相相比平均增加了0.87D,从而使DA1在水相中较在环己烷中具有更大的溶剂化效应,进一步说明在水相中构象具有更低的能量。
图4绘出了不同构象DA1分子在三相中的亲电反应指数ω随二面角φ变化情况,从纵向分析可知,DA1在气相中ω取值范围是0.036~0.038,在环己烷中的取值范围是0.032~0.034,在水中的取值范围是0.031~0.033,每相中的差值都是0.002,但变化规律不全相同,且三相中ω值都没有混交,即ωwater<ωcyclohexane<ωgas。由于N上的孤对电子不仅能形成分子内氢键,N-H键也可与极性大的水分子形成分子间氢键,同时酚羟基还可形成氢键,因而在极性溶剂中削弱了DA1的亲电性,在水环境中降低了其亲电反应活性,因此DA1在水中的构象最稳定。从横向分析可知,在150°≤φ≤210°时,三相中DA1的ω值都最小,此时DA1的亲电性最弱。

图5 TD-B3LYP/6-311++G(2 d,p)水平上DA1的振子强度f
2.2.2 DA1不同构象的UV-Vis光谱:用B3LYP方法对三相中DA1分子的UV-Vis光谱电子跃迁主要轨道、最大吸收波长和振子强度f进行了计算,结果表明水相中的振子强度高于油相环己烷和气相的,尤其是0°≤φ≤60°时,f的数值在0.11以上,气相中f数值在0.05~0.07范围之内,油相环己烷中则在0.05~0.09范围之内,两相中的f数值均不超过0.10。由此可知,水对DA1的UV-Vis能够产生显著的影响,使f数值增大。
不同溶剂对DA1的UV-Vis的影响不同,图5列出了三相中随二面角φ变化的f值。从图5可知,在同一相中,f随φ有规律的变化,不同相中,f值的变化规律是不同的。从表2中可知,在气相和环己烷中,当f数值最大时,λmax都在260 nm附近,而DA1的电子跃迁轨道发生了改变。在水相中,f数值在0.11以上,即0°≤φ≤60°时,λmax在210 nm附近,DA1的电子跃迁轨道发生了显著的变化,不是简单的HOMO→LUMO的跃迁,在60°<φ≤180°时,DA1的电子跃迁类型又变成了简单的HOMO→LUMO的跃迁,在180°~360°之间计算的结果是重复的,溶剂极性增加使UV-Vis光谱发生蓝移。
2.2.31H、13C-NMR分析:在0°≤φ≤360°范围内以每5°为步长进行旋转,用OPBE/6-311++G(2 d,p)//B3LYP/6-311++G(2 d,p)水平对已结构优化好的稳定的DA1分子进行1H、13C-NMR的研究,由1H、13C-NMR谱图可知,随着φ的转动,DA1中的H12、H17、H20、C4、C9、C10均发生明显的变化。0°≤φ≤180°范围内DA1在气相及两种溶剂中的相关1H、13C-NMR的化学位移计算值与实验值[29]比较可知,随着二面角的变化,上述关注的H和C原子的化学位移值在~0.7和~8 ppm内进行变化。

图6 OPBE/6-311++G(2 d,p)//B3LYP/6-311++G(2 d,p)水平上DA1中H17的化学位移值
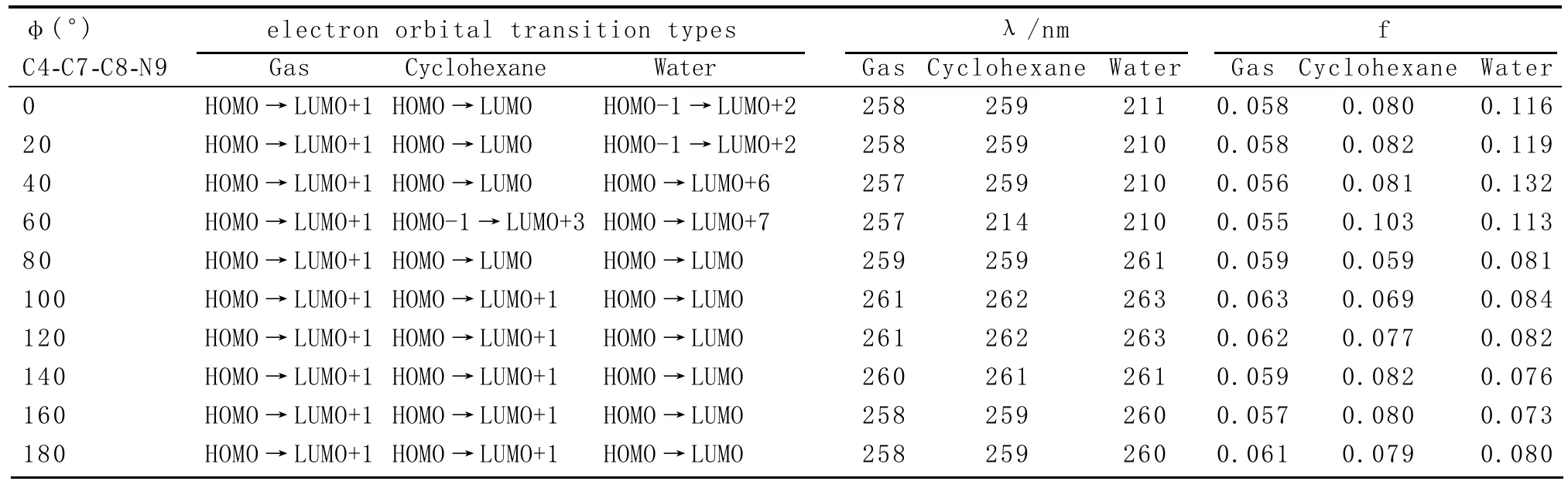
表2 DA1的UV-Vis计算值
不同溶剂对DA1中不同原子化学位移值的影响不同,图6绘出了0°≤φ≤360°范围内,DA1分子的不同构象在气相及两种溶剂中H17的化学位移变化值。从图6可知,H17的化学位移值在三相中的变化均呈“w”形,在0°~100°范围内,H17化学位移值逐渐减小,100°~180°范围内逐渐增大,但变化值随溶剂极性增加而逐渐降低,其他原子H20、H21、C4、C9、C10化学位移值呈类似状况。
分析比较1H、13C-NMR的化学位移计算值与实验测定值可知,当φ=0°时,所对应的C4、C9、C10在水相中的化学位移值与实验值之间的绝对误差Δ δ均最小。对三相中的1H、13C-NMR化学位移值与实验值之间的平均相对误差MRE进行计算,气态条件下,H12、H17、H20、C4、C9和C10分别为0.023、0.196、0.111、0.021、0.277、0.157;环己烷中,C4和C9的MRE为最小和最大值;水相中,C4的计算数据与实验值吻合最好。溶剂效应对1H-NMR的化学位移值产生显著的影响。随着溶剂极性的增大,1H-NMR的化学位移值增大,逐渐向低场移动,而13C-NMR的化学位移向高场移动。
3 结论
本研究采用杂化密度泛函方法B3LYP,在6-311++G(2 d,p)基组水平上对DA多种初始构象进行结构全优化,得到11种气相稳定结构,其中能量最低的是DA1,结合实验报道的振动光谱数据进行分析,证实了计算方法的可靠性。
对DA1进行了详细计算研究,在0°≤φ≤360°范围内,以5°旋转为步长,对DA1的72种构象异构体在气相、油相环己烷和水相中的构象变化和性质进行了计算。结果表明不同溶剂环境中,DA1构象转变难易程度不同,在0°≤φ≤60°范围内,随着溶剂极性增加而转动能垒升高,在60°≤φ≤180°范围则呈相反变化。势能变化曲线能用正弦函数较好拟合。溶剂极性增加,构象的极性μ也增加,而亲电反应指数ω则逐渐降低。运用TD-B3LYP/6-311++G(2 d,p)计算不同环境中DA1构象变化时的UV-Vis光谱,0°≤φ≤60°范围内的水相中吸收最大峰λmax在210 nm附近都不是HOMO与LUMO之间的电子跃迁,而气相中最大吸收峰都是HOMO与LUMO+1电子跃迁为主。在OPBE/6-311++G(2 d,p)//B3LYP/6-311++G(2 d,p)水平上对DA1在三相中的1H、13CNMR进行研究,溶剂效应对1H、13C-NMR的化学位移值产生显著的影响。随着溶剂极性的增大,1H和13CNMR的化学位移值分别向低场和高场移动。
[1] Owens NW, Braun C, Oneil JD, et a1. Effects of glycosylation of (2S, 4R)-4-Hydroxyproline on the conformation, kinetics and thermodynamics of prolylamide isomerization[J]. J Am Chem Soc, 2007, 129(38):11670-11671
[2] Hamelberg D, Shen T, Mccammon JA. Phosphoryl-ation effects on cis/trans isomerization and the backbone conformation of serine-proline motifs: accelerated molecular dynamics analysis[J]. J Am Chem Soc, 2005, 127(6):1969-1974.
[3] 张殿增. 量子药理学及其应用[J]. 自然杂志, 1988, 11(10):746-749, 769.
[4] Pine A, Shiner T, Seymour B, et a1. Dopamine, time and impulsivity in humans[J]. J Neurosci, 2010, 30(26):8888-8896.
[5] 曹磊, 罗红. 小剂量多巴胺联合前列地尔治疗慢性肾功能不全临床研究[J]. 淮海医药, 2012, 30(1):31-33.
[6] 杨丽娟, 杨晓春, 袁玉芳. 硫酸镁联合多巴胺治疗新生儿缺氧缺血性脑病的临床研究[J]. 徐州医学院学报, 2010, 30(2):118-120.
[7] He PG, Yu YL, Fang YZ. Determination of neurotransmit-ter dopamine in the presence of ascorbic acid using lipoic acid coated electrochemically pretreated carbon fibre microelectrode[J]. Chinese J Anal Chem, 1996, 24(4):407-410.
[8] Xu JJ, Wang Y, Fang HQ, et al. Electrochemical behaviours and amperometric determination of dopamine at gold electrode modified by thionine covalenyly bound to selfassembled monolayers[J]. Chinese J Anal Chem, 1998, 26(4):428-430.
[9] Lin XQ, Lu LP, Jiang XH. Voltammetric behavior of dopamine at ct-DNA modified carbon fiber micro-electrode[J].Microchim Acta, 2003, 143(4):229-235.
[10]Hu C, Zhang Y, Bao G, et al. DNA functionalized singlewalled carbon nanotubes for electrochemical detection[J]. J Phys Chem B, 2005, 109(43):20072-20076.
[11] 吴莹, 范瑞溪, 狄俊伟. 多巴胺在液/液界面上电子传递的电化学研究[J]. 分析化学, 1996, 24(8):873-876.
[12]Solmajer P, Kocjan D, Solmajer T. Conformational study of catecholamines in solution[J]. Z Naturforsch, C: Biosci, 1983,38(9-10):758-762.
[13] 李志锋, 李会学, 唐慧安, 等. 多巴胺DA分子的构象异构及其一水复合物的结构与性质[J]. 原子与分子物理学报, 2010,27(2):226-232.
[14]Urban JJ, Cramer CJ, Famini GR. A computational study of solvent effects on the conformation of dopamine[J]. J Am Chem Soc, 1992, 114(21):8226-8231.
[15]Cramer CJ, Truhlar DG. General parameterized SCF model for free energies of solvation in aqueous[J]. J Am Chem Soc,1991, 113(22):8305-8311.
[16] 张荣, 罗三来, 郑敦胜. 生物分子溶液中的弱相互作用研究进展[J]. 化学研究, 2008, 19(1):102-105.
[17] 李宝宗. 可乐定分子构象异构和互变异构的理论研究[J]. 化学学报, 2006, 64(4):278-282.
[18]Barone V, Bloino J, Biczysko M. Validation of the DFT/N07D computation model on the magnetic, vibrational and electronic properties of vinyl radical[J]. Phys Chem Chem Phys, 2010, 12:1092-1101.
[19]Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density[J]. Phys Rev, 1988, 37(2):785-789.
[20] Stephens PJ, Devlin FJ, Chabalowski CF, et al. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force field[J]. Phys Chem,1994, 98(45):11623-11627.
[21]Wu A, Zhang Y, Xu X, et al. Systematic studies on the computation of nuclear magnetic resonance shielding constants and chemical shifts: the density functional models[J].J Comput Chem, 2007, 28(15):2431-2442.
[22]Parrr G, Yang W. Density functional theory of atoms and molecules[M]. Oxford: Oxford University Press, 1989.
[23] Mulliken RS. A new electroaffinity scale together with data on valence states and on valence ionization potentials and electron affinities[J]. J Chem Phys, 1934, 2:782-793.
[24]Ayers PW, Parr RG, Pearson RG. Elucidating the hard/soft acid/base principle: A perspective based on half-reactions[J]. J Chem Phys, 2006, 124(19):194107-194108.
[25]Hohenberg P, Kohn W. Inhomogeneous electron gas[J]. Phys Rev, 1964, 136(3B):B864-B871.
[26]Kohn W, Sham LJ. Self-consistent equations including exchange and correlation effects[J]. Phys Rev, 1965, 140(4A):A1133-A1138.
[27]Pople JA, Gill PMW, Johnson BG. The performance of a family of density functional methods[J]. Chem Phys Lett,1992, 199:557.
[28]Becke AD. Density-functional thermochemistry. III. the role of exact exchange[J]. J Chem Phys, 1993, 98(7):5648.
[29]Sivakumar R, Divakar S. Enzymatic syntheses of dopamine glycosides[J]. Enzyme Microb Technol, 2009, 44(1):33-39.
[30]Merrick JP, Moran D, Radom L. An evaluation of harmonic vibrational frequency scale factors[J]. J Phys Chem A, 2007,111(45):11683-11700.
猜你喜欢
化工管理(2021年7期)2021-05-13 00:45:22
环境保护与循环经济(2020年4期)2020-06-08 10:43:48
石油地质与工程(2019年3期)2019-09-10 08:27:54
中国特种设备安全(2019年1期)2019-03-13 01:06:28
中国钼业(2019年2期)2019-01-19 15:54:06
水利技术监督(2016年6期)2017-01-15 14:01:33
温州大学学报(自然科学版)(2016年1期)2016-10-27 14:57:48
应用化工(2014年7期)2014-08-09 09:20:23
湿法冶金(2014年3期)2014-04-08 01:04:51
无机化学学报(2014年5期)2014-02-28 17:31:40
