
二联咪唑环芳识别氨基酸甲酯的理论研究
2013-01-04 06:09刘祥均娄永峰杨明理向明礼
成都工业学院学报 2013年1期
刘祥均,娄永峰,肖 蓉,杨明理,向明礼
(四川大学 华西医院生物治疗国家重点实验室,成都 610041)
1949年,Brown和Farthing[1]从二甲苯的低压热解聚合物中分离出了第一个环芳化合物。在用X射线晶体衍射的方法解析后,发现这是一个由两段次乙基桥把2个苯环以“面对面”的方式从对位连接起来的全新化合物,并将其命名为“二对二甲苯”[1],如图1所示。这类含有芳香环的环状有机化合物[2]由于可能存在某些诱人的特性[3]而备受关注。自被发现[1]以来,环芳被广泛应用于手性识别[4-5]、超分子[6]、分子催化[7]、抗菌[8]等领域。咪唑环芳中的咪唑,既能作为质子受体又能作为质子供体,具有特殊亲核、亲电性[9]。咪唑也是生物体系中的重要基团[10]。不仅如此,近年来的研究还发现含咪唑环的化合物有的具有抗炎活性[11],有的成为新型抗癌制剂的先导化合物[12],有的能抑制表皮生长因子受体(EGFR)的酶活性[13]。因而,咪唑环芳成为环芳研究[9,14]中的一个热点。
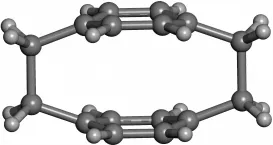
图1 “二对二甲苯”的立体维结构
鉴于环芳识别氨基酸甲酯的理论研究并不多见,本文拟借助密度泛函理论(DFT),对新近合成的一个二联咪唑环芳受体[9](如图2所示)与一系列氨基酸甲酯配体间的相互作用,从理论上进行探讨,以期在更细微的层面对其获得更深入的认识。
1 计算方法
如图2所示,二联咪唑环芳(DBICP)是由2条1,2-二乙氧基乙烷链将2个2,2’-联咪唑连接在一起构成的。DBICP所识别的5个氨基酸甲酯(AME)配体分别为缬氨酸甲酯(VOM)、脯氨酸甲酯(POM)、亮氨酸甲酯(LOM)、甘氨酸甲酯(GOM)和丙氨酸甲酯(AOM)。计算工作分为3部分:1)是获得DBICP与AME所形成的复合物的构象;2)对复合物以及构成复合物的各单体作NBO分析;3)是计算构成复合物的各单体的溶剂可及面积。

图2 二联咪唑环芳受体的二维结构
对于DBICP受体及其配体的单体三维结构,采用MarvinSketch(http://www.chemaxon.com)构建并用Gaussian 03[15]在B3LYP/6-31G(d)理论水平上进行优化得到。而DBICP受体与AME所形成的复合物的初始构象,则采用基于模拟退火的分子动力学程序CDOCKER[16],将AME配体与DBICP受体对接。对所产生的随机构象进行逐渐升温和模拟退火动力学模拟。对接时,选用CHARMM力场。将受体的几何中心与所设置的结合位点的几何中心重叠。球半径参数设置为5 A。。每个配体与受体对接所产生的随机构象的最大数目置为1 000。对接和动力学模拟过程中的其余参数采用默认值。
对于每一个复合物,按能量高低顺序将经过动力学模拟后得到的每种复合物的构象排序,从中选出10个能量值为负值的最低能量构象。然后用Turbomole[17]程序,在BLYP/def-TZVP[18]水平进行优化。最后从优化收敛后的结果中,选出能量最低的构象作为该复合物的优化构象,做进一步的NBO分析。对于复合物,将优化收敛后得到的最低能量构象在相同理论和基组下作NBO分析。对于之前经结构优化后得到的DBICP受体和AME配体,也在BLYP/def-TZVP[18]水平进行优化并作NBO分析。
构成复合物的各单体的溶剂可及面积,用MarvinSketch在pH值为7.4、溶剂半径为1.4 A。条件下,分别计算其带正电荷原子的溶剂可及表面积SASA+和带负电荷的原子的溶剂可及表面积SASA-。

表1 溶液酸碱性对3种偶氮化合物颜色的影响
2 结果与讨论
2.1 结合常数的计算
通常认为,复合物的最高占据轨道能级、最低空轨道能级、形成复合物过程中受体与配体间所转移的电荷量参数等是衡量复合物稳定性的重要指标。表1列出了脯氨酸甲酯单体及在DBICP-POM复合物中的NBO电荷分布以及形成复合物前后的电荷分布变化。表2中反映了各AME在与DBICP形成复合物后的电荷转移量。
但有研究表明[19],咪唑环芳对氨基酸甲酯的识别比较复杂,上述各计算量甚至受-配体间的相互作用能等,都与实验测定的复合物结合常数之间没有简单的对应关系。因此对于这类复合物的理论研究,需考虑新的因素。笔者注意到[20-21]配体的溶剂可及表面积SASA在分子识别过程中所具有的重要地位。它与溶剂化的非极性贡献有关。而这一点,在量子化学计算结果中难以准确体现[22]。因此,在探讨如何从理论上计算DBICP-AME复合物的结合常数时,除受、配体间的电荷转移量外,SASA被重点考虑。

表2 复合物结合常数K的实验值[9]、计算值等相关重要参数
笼统地考虑SASA,可能会掩盖某些重要信息。为此,将SASA进一步细分为带正电的原子的溶剂可及表面积SASA+和带负电的原子的溶剂可及表面积SASA-。DBICP 受体的 SASA+=520.02 A。2,SASA-=203.14 A。2。AME 配体的相应计算结果见表2。
为了从理论上计算DBICP识别AME形成复合物的结合常数,笔者构建了如下2参数结合常数定量计算模型:

式中α、β和γ为待定参数。用表2中的数据,对(1)式进行多项式回归最小二乘拟合求解,得:

将复合物结合常数的计算值(见表2)与实验值[9]作图。如图3所示,计算值与实验值间有良好的相关性,相关系数高达0.997。
2.2 相互作用分析
从表2可知,复合物的结合常数与AME配体带负电原子的溶剂可及表面积SASA-有一定的相关性。而以此参数结合受—配体间的电荷转移量所建立的复合物结合常数的定量计算模型,又能很准确地表达复合物受—配体间的结合程度。这就启发我们在探讨DBICP受体与AME配体间的相互作用时,需重点考虑AME配体的带负电原子的溶剂可及表面积。

图3 复合物结合常数实验值[9]与计算值的相关图

图4 受体分子的NBO电荷分布图
受—配体间的非键相互作用,本质上仍属于静电相互作用的范畴。因此,首先考察一下DBICP受体的电荷分布。根据NBO电荷分析的结果,将DBICP受体的电荷分布作出如图4所示的电荷分布图。图中带负电荷的原子用球状表示,用圆球表示的8个N原子,4个氧原子和4个直接与N原子相连接的C原子明显地带负电。如果把DBICP受体带负电荷的区域分成4块的话,从中可以直观地看出2点特征:1)每块都是由联咪唑环的2个同侧N原子,1个与N原子相连接的C原子和1个O原子构成(参见图4中的虚线椭圆区域);2)在DBICP受体分子中,带负电荷的原子相对集中,而带正电荷的原子则相对分散。
由于受体分子负电荷分布相对集中,而静电相互作用又属于长程(long range)相互作用,因此AME配体分子的带负电荷原子与受体分子中带负电荷原子间的排斥相互作用,对复合物的稳定性结合产生至关重要的影响。所以总体上讲,配体分子的SASA-越小,配体与受体间的排斥作用就越小,对复合物的稳定性贡献就越大。在5个配体分子中,POM的SASA-最小(见表2),DBICP-POM的结合常数最大,复合物最稳定;VOM和GOM的SASA-较大,它们对应的复合物的稳定性较差;而LOM和AOM的SASA-属于最大的那类,它们与BDICP形成的相应的复合物,稳定性最差。这与实验测定结果是一致的。
各配体分子与DBICP受体间最低能量构象,如图5所示。作用模式各有千秋,但有2个共同点:1)在受—配体间的相互作用中,都观察到氢键相互作用(氢键供体与受体间的距离,见表2);2)配体采取的是一种既与受体形成氢键,又尽可能减小其带负电区域与受体带负电荷区域间的排斥作用的模式。与受体发生相互作用,都是以其氮端朝向受体分子。
VOM、LOM和GOM与DBICP间有相似的作用模式。其带负电的氮端大体上都朝向受体分子中负电荷较集中的其中一块区域(联咪唑同侧的2个N原子,1个与N原子相连接的C原子和1个O原子)。距离其带负电氮端最近的,是咪唑环上的N原子。相较于VOM和GOM的酯端都尽量远离受体,LOM带负电的酯端靠向受体带负电的咪唑环。因而LOM与受体间有较大的静电排斥作用。
POM和AOM与DBICP间的作用模式较为特殊。在DBICP分子中,与POM的带负电的氮端靠得较近的负电荷原子,只有1个咪唑上的N原子,2个与N原子相连接的C原子和1个O原子。而距离POM带负电的氮端最近的,不是咪唑环上的N原子,而是咪唑环上的一个带弱负电的碳原子。这种模式使得POM与DBICP间形成的氢键较弱(见表2),但同时,它们间的负—负排斥作用最小。在AOM与DBICP间的相互作用模式中,AOM的带负电的氮端所靠近的负电荷原子,不是咪唑环上的带负电的N原子或碳原子,而是1,2-二乙氧基乙烷链上的O原子。AOM带负电的酯端也没有远离受体,而是靠向带负电的与咪唑环相连的C原子。因此,AOM与DBICP受体间也有较强的静电排斥作用,它们形成的复合物稳定性较差。
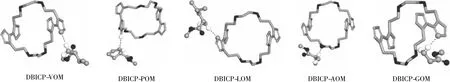
图5 二联咪唑环芳与AMEs间的相互作用模式
3 结语
为研究二联咪唑环芳(DBICP)对一系列氨基酸甲酯(AME)的识别作用,首先用MarvinSketch构建了受体和配体分子单体,并用Gaussian 03在B3LYP/6-31G(d)水平上优化结构,得到了各单体的三维结构。然后将结构优化后的每一个AME配体,都用基于模拟退火的分子动力学程序CDOCKER对接到DBICP受体。为每个复合物随机生成的1 000个构象,经动力学模拟和能量排序后,选出10个能量最低构象作为复合物的初始构象。再用Turbomole在BLYP/def-TZVP水平对构成复合物的单体及每个复合物的初始构象进行几何构型优化,将收敛后得到的最低能量构象作为复合物的结构。最后在相同理论和基组下作NBO分析。
鉴于咪唑环芳对氨基酸甲酯的识别的复杂性和配体的溶剂可及表面积SASA在分子识别过程中所具有的重要地位,把AME配体的SASA进一步表达为带负电荷原子的溶剂可及表面积SASA-和带正电荷原子的溶剂可及表面积SASA+。发现配体分子的SASA-是影响DBICP识别AME的主要因素。用受配体间的电荷转移量及配体SASA-构建了表达复合物稳定性的二参数定量关系模型。该模型能准确地计算DBICP-AME复合物的结合常数,计算值与实验值间的相关系数高达0.997。
受、配体间的相互作用模式分析表明,各AME配体与DBICP受体间的相互作用态势千差万别的同时,也表现出一些共同特征:在受—配体间都观察到了氢键相互作用;配体采取的是一种既与受体形成氢键,又尽可能减小其带负电区域与受体带负电荷区域间的排斥作用的模式。与受体发生相互作用,都是以其氮端朝向受体分子。
[1]BROWN C J,FARTHING A C.Preparation and Structure of Di-p-Xylylene[J].Nature,1949(164):915 -916.
[2]CRAM D J,CRAM J M.Cyclophane chemistry:bent and battered benzene rings[J].Acc Chem Res,1971,4(6):204 - 213.
[3]CRAM D J,STEINBERG H.Macro rings.I.preparation and spectra of the paracyclophanes[J].J Am Chem Soc,1951,73(12):5691 -5704.
[4]PU L.Enantioselective fluorescent sensors:a tale of bINOL[J].Acc Chem Res,2011,:DOI:10.1021/ar200048d.
[5]YANG L,QIN S,SU X,et al.1,1'-Binaphthyl-based imidazolium chemosensors for highly selective recognition of tryptophan in aqueous solutions[J].Org Biomol Chem,2010,8(2):339 -348.
[6]CAMACHO D H,SALO E V,ZILLER J W,et al.Cyclophane-based highly active late-transition-metal catalysts for ethylene polymerization[J].Angew Chem Int Ed,2004(43)1821 -1825.
[7]POYATOS M,MATA J A,PERIS E.Complexes with poly(N-heterocyclic carbene)ligands:structural features and catalytic applications[J].Chem Rev,2009,109(8):3677 -3707.
[8]MELAIYE A,SUN Z,HINDI K,et al.Silver(I)-midazole cyclophane gem-diol complexes encapsulated by electrospun tecophilic nanofibers:formation of nanosilver particles and antimicrobial activity[J].J Am Chem Soc,2005,127(7):2285 -2291.
[9]XU X,WANG X,WU A,et al.Synthesis and molecular recognition of novel multiimidazole cyclophanes[J].J Heterocyclic Chem,2009,46(6):1137 - 1141.
[10]YUAN Y,GAO G,JIANG Z,et al.Synthesis and selective anion recognition of imidazolium cyclophanes[J].Tetrahedron,2002,58(44):8993 -8999.
[11]HERN NDEZ P,CABRERA M,LAVAGGI M L,et al.Discovery of new orally effective analgesic and anti-inflammatory hybrid furoxanyl N-acylhydrazone derivatives[J].Bioorg Med Chem,2012,20(6):2158 -2171.
[12]SUBTEL'NA I,ATAMANYUK D,SZYMAN SKA E,et al.Synthesis of 5-arylidene-2-amino-4-azolones and evaluation of their anticancer activity[J].Bioorg Med Chem,2010,18(14):5090 -5102.
[13]ABOU-SERI S M,FARAG N A,HASSAN G S.Novel diphenylamine 2,4'-dicarboxamide based azoles as potential epidermal growth factor receptor inhibitors:synthesis and biological activity[J].Chem Pharm Bulletin,2011,59(9):1124 -1132.
[14]GABUTTI S,SCHAFFNER S,NEUBURGER M,et al.Planar chiral asymmetric naphthalenediimide cyclophanes:synthesis,characterization and tunable FRET properties[J].Org Biomol Chem,2009(7):3222 - 3229.
[15]FRISCH M J,TRUCKS G W,SCHLEGE H B,et al.Gaussian 03,Revision C.02[Z].Wallingford CT:Gaussian Inc,2004.
[16]WU G,ROBERTSON D,BROOKS C,et al.Detailed analysis of grid-based molecular docking:a case study of CDOCKER-A CHARMm-based MD docking algorithm[J].J Comput Chem,2003,24(13):1549 -1562.
[17]TREUTLER O,AHLRICHS R.Efficient molecular numerical integration schemes[J].J Chem Phys,1995(102):346 -354.
[18]EICHKORN K,TREUTLER O,OEHM H,et al.Auxiliary basis sets to approximate Coulomb potentials[J].Chem Phys Lett,1995,240(4):283 -289.
[19]娄永峰,肖蓉,向明礼,等.新型多苯并咪唑环芳识别氨基酸甲酯的分子动力学和密度泛函研究[J].成都电子机械高等专科学校学报,2012,15(4):10 -15.
[20]ZHOU Z,MADURA J D.Relative free energy of binding and binding mode calculations of HIV‐1 RT inhibitors based on dock‐MM‐PB/GS[J].Proteins:Structure,Function,and Bioinformatics,2004,57(3):493 -503.
[21]BONNET P,BRYCE R A.Molecular dynamics and free energy analysis of neuraminidase-ligand interactions[J].Protein Sci,2004,13(4):946 -957.
[22]XIANG M,LIN Y,HE G,et al.Correlation between biological activity and binding energy in systems of integrin with cyclic RGD-containing binders:a QM/MM molecular dynamics study[J].J Mol Model,2012,18(11):4917 -4927.
猜你喜欢
中国民间疗法(2021年17期)2021-11-04
云南化工(2020年11期)2021-01-14
唐山师范学院学报(2020年6期)2020-04-16
温州大学学报(自然科学版)(2016年1期)2016-10-27
西安工程大学学报(2016年3期)2016-06-05
中国卫生标准管理(2015年2期)2016-01-14
化工进展(2015年3期)2015-11-11
中国当代医药(2015年10期)2015-03-01
海军医学杂志(2015年2期)2015-02-27
西南军医(2015年2期)2015-01-22
