
外源性脑源性神经营养因子对Alzheimer大鼠行为学的影响及其作用机制
2012-12-03 08:09:12徐爱华王维商秀丽
中国医科大学学报 2012年12期
徐爱华,王维,商秀丽
(中国医科大学附属第一医院1.康复医学科;2.神经内科,沈阳110001)
阿尔茨海默病(Alzheimer′s disease,AD)是一种以进行性认知功能减退为特点的神经系统变性疾病。记忆障碍,尤其是近记忆障碍,是AD典型的首发征象,随后出现远记忆受损、时间及空间定向障碍,晚期多伴有人格改变,严重影响老年人的生活质量。在所有与年龄相关的痴呆中,AD约占50%[1]。随着社会老龄化的发展,估计到2050年我国65岁及以上老人约占总人口的35%,80岁及以上老人约占22%,AD患者预期可达2500万,将成为影响家庭和社会发展的一个重要制约因素,因此必须对其进行积极防治。脑源性神经营养因子(brain-derived neurotrophic factor,BDNF)是脑内广泛存在的一种神经营养因子。海马CA3区是与学习和记忆密切相关的脑功能区,也是BDNF及其受体TrkB在脑内表达最集中的部位[2]。研究发现,AD患者脑内BDNF mRNA及蛋白表达较正常人脑明显降低[3],同时相应脑区突触素的表达也明显降低,说明二者之间可能存在着某种联系,共同在AD发病中起作用。因此,我们通过对大鼠海马CA3区立体定位注射冈田酸(okadaic acid,OA)建立模拟AD的大鼠模型,再用外源性BDNF对其作用,观察外源性BDNF对脑内突触素表达的影响以及对大鼠行为学的作用,探讨其可能的作用机制。
1 材料与方法
1.1 材料
1.1.1 实验动物:健康成年雄性Wistar大鼠60只,体质量200~250 g,购自沈阳医学院实验动物中心。饲养温度(20±2)℃,湿度70%,自然通风,自然光照,大鼠可自由接触食水。
1.1.2 实验试剂:OA(美国Upstate公司),BDNF(美国 Chemicon公司),突触素抗体、K252a、LY294002、pThr231抗体(美国Santa Cruz公司)。
1.2 实验方法
1.2.1 实验动物分组:60只大鼠随机平均分为正常对照组、假手术组、OA组、BDNF组、K252a组和LY294002组。
1.2.2 动物模型制作:大鼠用10%水合氯醛300 mg/kg腹腔内注射麻醉,俯卧位固定于大鼠脑立体定位仪上,沿颅顶正中线切开皮肤,暴露头骨,按照大鼠脑立体定位图谱定位:AP-3.8 mm,ML±3.8 mm,DV 4.0 mm,即为海马CA3区注射点。假手术组大鼠每侧海马注射2 μL生理盐水;OA组每侧海马注射2μL OA(0.2 μmol/L);BDNF 组每侧注射2μL OA+2 μL BDNF(50 ng/mL);K252a 组每侧注射2μL OA+2 μL BDNF+2 μL K252a (0.2 μmol/L);LY294002 组每侧注射2μL OA+2 μL BDNF+2 μL LY294002(0.2 μmol/L)。
1.2.3 水迷宫实验
1.2.3.1 定位航行试验 历时4 d,每天上、下午各2次,将大鼠面向水迷宫池壁分别从4个入水点放入水中,记录大鼠找到平台所需要的时间,即逃避潜伏期。如大鼠在2 min内找不到平台,则由试验者将其引向平台,其潜伏期为120 s。每次训练间隔为60 s。
1.2.3.2 空间探索试验 定位航行试验结束后撤除平台,然后将鼠任选一个点放入水中,测其2 min内跨越原平台所在位置的次数及游泳轨迹。
1.2.4 标本采集:用于免疫组化检测的动物用10%水合氯醛300 mg/kg经腹腔注射进行麻醉,然后开胸暴露心脏,先用生理盐水经左室心尖插管快速灌注,再用4%多聚甲醛溶液(pH 7.2~7.4)持续灌注约300 mL,至大鼠四肢变硬后断头取脑,并于4%多聚甲醛中后固定48 h。用于Western blot检测的动物,用水合氯醛深度麻醉后直接断头取脑,于冰盒上快速分离双侧海马组织,放入1.5 mL Eppendorf管中,立即-80℃冻存。
1.2.5 Western blot:将冻存的组织融化。4℃裂解细胞,离心15 min,取上清。应用酚试剂法测定样本中蛋白浓度。将电泳后的硝酸纤维素膜在转印液中浸泡数分钟后,转印2 h。用含10%脱脂奶粉的封闭液封闭,4℃过夜。将封闭过夜的膜取出,加入p38(1∶1000)及 pThr231(1∶1000),室温 4 h。将一抗孵育后的膜用TTBS洗5 min,2次,然后加入HRP标记的二抗(1∶2000),室温2 h。二抗洗膜后,在暗室将荧光剂A液B液等量混匀后,立即加到膜上,反应1 min,将膜固定到曝光盒中,曝光1~5 min,依次显影定影,比照曝光板上的膜记录下marker和各条带的位置。将蛋白印记显影图扫描,利用图像分析软件Scion Image对蛋白带进行光密度分析。以β-actin作为内对照,将每一个蛋白带的光密度值与相应内对照β-actin蛋白带的比值作为所测蛋白的半定量指标。
1.3 统计学处理
2 结果
2.1 定向航行试验
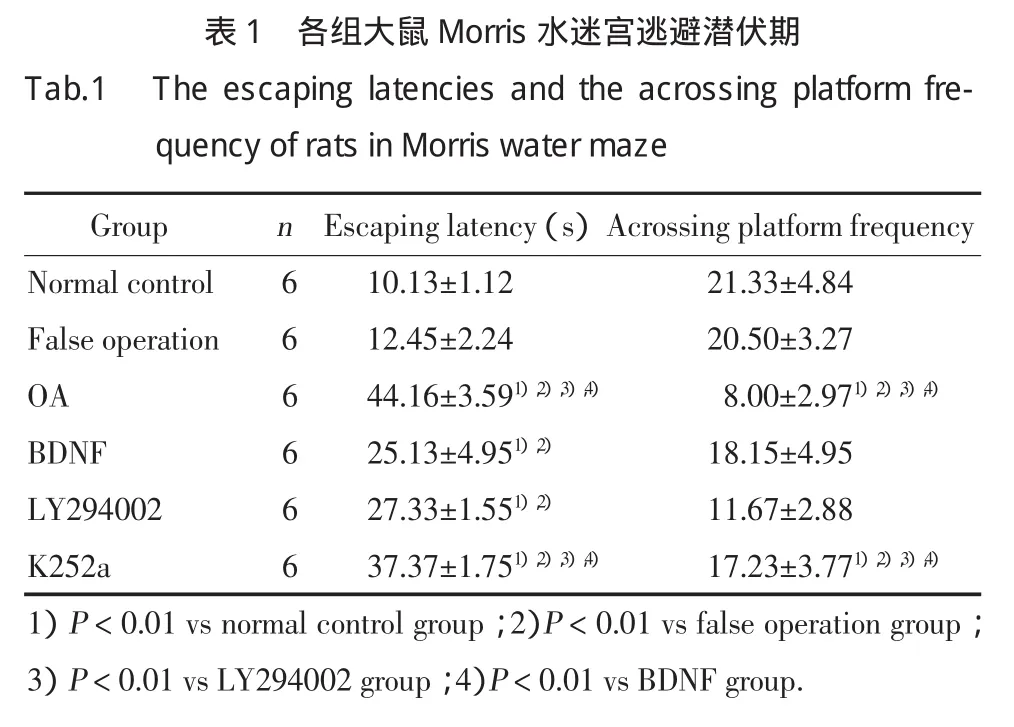
OA组及K252a组大鼠逃避潜伏期较正常组、假手术组、LY294002组及BDNF组明显延长(P<0.01);BDNF及LY294002组大鼠逃避潜伏期较正常组及假手术组也明显延长(P<0.01)。见表1,图1。

2.2 空间探索试验
OA组及K252a组大鼠跨越平台次数较正常对照组、假手术组、LY294002组及BDNF组明显减少(P<0.01);BDNF 组、LY294002组、正常对照组及假手术组比较,大鼠跨越平台次数的差异无统计学意义(P>0.05)。见表1,图2。

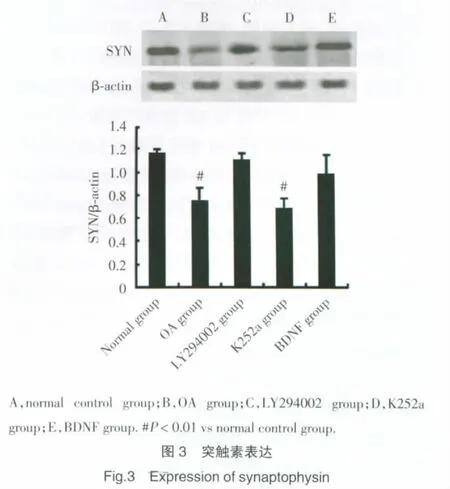
2.3 突触素表达
正常对照组、BDNF组及LY294002组突触素表达较OA组和K252a组增高(P<0.01),但正常对照组、BDNF组及LY294002组3组之间无统计学差异(P>0.05)。见图3。
2.4 pThr231表达
OA组、LY294002组和K252a组pThr231表达较正常对照组及BDNF组明显增多(P<0.01),但OA组、LY294002组及K252a3组3组比较无统计学差异(P>0.05);BDNF组与正常对照组比较无统计学差异(P>0.05)。见图4。

3 讨论
突触作为神经系统的特征性组织结构之一,是神经元之间相互作用的基本结构单位,是记忆形成的结构基础。研究表明,突触丢失是AD早期脑内一个重要的病理学特征[4]。突触素是突触终末特异性标记物,构成突触囊泡特异性膜通道,参与囊泡的转运与排放。突触素密度与AD痴呆严重程度呈负相关[5]。我们在实验中用OA对大鼠海马CA3区进行立体定位注射,引起了大鼠显著的空间记忆障碍,海马突触素蛋白及其mRNA表达明显减少,tau蛋白Thr231位点过磷酸化。我们认为大鼠空间记忆障碍主要是由突触结构及功能异常引起的,而tau蛋白的过磷酸化及神经营养因子的缺乏是引起突触结构功能异常的主要原因。一方面,OA可直接导致tau过磷酸化;另一方面,BDNF及其受体的缺乏可引起PI-3K/AKT通路的失活,GSK-3β表达上调,增加tau蛋白的过磷酸化[6]。过磷酸化的tau蛋白可引起微管聚合、组装、稳定性及功能异常,从而可导致突触形成、生长、囊泡转运等多方面障碍,使得突触素表达减少。突触素表达的减少意味着突触数量的减少、突触囊泡转运能力的下降、突触传递功能的减弱,由此可引起神经系统信息传递、加工和储存障碍,导致学习记忆功能减退。
许多研究证明,BDNF通过对突触生长、突触可塑性及突触传递的调节作用,在认知、学习和记忆形成中起重要作用[7]。BDNF在突触可塑性的突触前和突触后机制中都是必需的,并且对记忆形成所必需的长时程增强的形成及保留[8]都是必需的。在细胞水平,BDNF被证明可增加突触前神经递质的释放,调节现有的突触运输,并能增加突触形成[9];在分子水平,BDNF增加β-连环蛋白酪氨酸残基的磷酸化,减少钙黏蛋白-β-连环蛋白黏着复合物的解离[10],促进细胞内环境的稳定。本研究应用外源性BDNF后,tau蛋白Thr231位点过磷酸化减少,海马突触素蛋白及其mRNA表达明显升高,大鼠空间认知明显改善。说明外源性BDNF确实可通过减少tau蛋白磷酸化、增加突触形成、改善突触结构和功能等多重途径在记忆形成和保留过程中起着重要作用。
我们将TrkB受体抑制剂K252a与BDNF同时海马内注射,BDNF的所有作用都被减弱,说明BDNF的生物学效应是以其受体TrkB为基础的。BDNF与TrkB受体结合后将其激活,后者又可通过自动磷酸化引起MAPK、PI-3K等通路的激活[11]。研究证明,TrkB在调节海马齿状回神经元突触结构中起着重要作用,特异敲除TrkB的小鼠突触前及突触后结构都发生了改变,共同影响皮质对齿状回细胞的联合输入,并对CA3区的锥体细胞产生负反馈抑制[12]。研究中我们发现,PI-3K抑制剂LY294002存在时BDNF使tau蛋白去磷酸化作用减弱,但BDNF提高突触素表达及其对大鼠行为学的改善并没受影响,说明这一作用是不依赖于或不仅依赖于PI-3K/AKT通路的。
[1]Lorke DE,Wai MS,Liang Y,et al.TUNEL and growth factor expression in the prefrontal cortex of Alzheimer patients over 80 years old[J].Int J Immunopathol Pharmacol,2010,23(1):13-23.
[2]Sylvie B,Karim B,Laetitia T,et al.Hippocampal BDNF expression in a Tau transgenic mouse model [J].Curr Alzheimer Res,2012,9(4):406-410.
[3]Chen Q,Zhou Z,Zhang L,et al.Tau protein is involved in morphological plasticity in hippocampal neurons in response to BDNF[J].Neurochem Int,2012,60(3):233-242.
[4]Coleman PD,Yao PJ.Synaptic slaughter in Alzheimer′s disease[J].Neurobiol Aging,2003,24(8):1023-1027.
[5]Pontén E,Fredriksson A,Gordh T,et al.Neonatal exposure to propofol affects BDNF but not CaMKII,GAP-43,synaptophysin and tau in the neonatal brain and causes an altered behavioural response to diazepamintheadultmousebrain[J].BehavBrain Res,2011,223(1):75-80.
[6]Elliott E,Atlas R,Lange A,et al.Brain-derived neurotrophic factor induces a rapid dephosphorylation of tau protein through a PI-3 kinase signalling mechanism[J].Eur J Neurosci,2005,22(5):1081-1089.
[7]SchindowsdiK,BelarbiK,BueeL.NeurotrophicfactorsinAlzheimer′s disease:role of axonal transport[J].Genes Brain Behav,2008,7(1):43-56.
[8]Bekinschtein P,Cammarota M,Igaz LM,et al.Persistence of longterm memory storage requires a late protein synthesis-and BDNF dependent phase in the hippocampus[J].Neuron,2007,53(2):261-277.
[9]Tyler WJ,Pozzo-Miller LD.BDNF enhances quantal neurotransmitter releaseandincreasesthenumberof docked vesicles at the active zones of hippocampal excitatory synapses[J].J Neurosci,2001,21(12):4249-4258.
[10]Bamji SX,Rico B,Kimes N.BDNF mobilizes synaptic vesicles and enhances synapse formation by disrupting cadherin-beta-catenin interactions[J].J Cell Biol,2006,174(2):289-299.
[11]Zhang F,Kang Z,Li W,et al.Roles of brain derived neurotrophic factor/tropomyosin-related kinase B (BDNF/TrkB)signalling in Alzheimer′s disease[J].J Clin Neurosci,2012,19(7):946-949.
[12]Steve CD,Robert JK,Cynthia W,et al.Altered morphology of hippocampal dentate granule cell presynaptic and postsynaptic terminalsfollowingconditionaldeletionofTrkB[J].Hippocampus,2008,18(7):668-678.
猜你喜欢
作文周刊·小学二年级版(2022年20期)2022-05-05 01:33:06
创新作文(小学版)(2019年10期)2019-09-25 08:12:28
天津医科大学学报(2019年6期)2019-08-13 07:04:42
国际呼吸杂志(2019年8期)2019-04-29 09:15:10
中成药(2017年9期)2017-12-19 13:34:56
小学生学习指导(低年级)(2017年5期)2017-05-04 04:14:38
吉林大学学报(医学版)(2015年1期)2015-12-17 07:47:22
安徽医科大学学报(2015年9期)2015-12-16 11:09:42
作文与考试·小学高年级版(2015年17期)2015-05-30 10:48:04
遗传(2014年3期)2014-02-28 20:59:01
