
水溶液中碳酸铀酰化合物的电子结构
2012-11-30 10:41:24辜家芳陆春海陈文凯章永凡
物理化学学报 2012年4期
辜家芳 陆春海 陈文凯,* 陈 勇 许 可 黄 昕 章永凡
(1福州大学化学系,福州350108;2成都理工大学核技术与自动化工程学院,成都610059)
水溶液中碳酸铀酰化合物的电子结构
辜家芳1陆春海2陈文凯1,*陈 勇1许 可1黄 昕1章永凡1
(1福州大学化学系,福州350108;2成都理工大学核技术与自动化工程学院,成都610059)
应用相对论密度泛函理论系统研究了水溶液中非水合化和水合化碳酸铀酰化合物Cn/m(其中n和m分别为结构中碳酸配体和水配体的个数)的结构.溶剂效应采用类导体屏蔽模型(COSMO),并采用零级规整近似(ZORA)方法考虑标量相对论效应和旋-轨耦合相对论效应.电子跃迁采用包含旋-轨耦合相对论效应的含时密度泛函理论并在相关交换势中采用轨道势能统计平均(SAOP)做近似计算.结果表明碳酸配体对配合物结构和电子跃迁有很大的影响.C3/0配合物的稳定性可归于5f轨道参与了高占据轨道的成键作用.增加碳酸盐配体导致最大波长的蓝移,并在近可见光区域出现高强度的吸收.
铀酰;UV-Vis;溶剂效应;含时密度泛函理论;旋-轨耦合相对论效应
1 Introduction
The uranyl ion is easy to interact with carbonate ligands to form complexesand plays an important role in migration from a nuclear waste repository or in ac-cidental site contamination in natural water.Hence,to understand the properties of the uranyl carbonate complexes in environment is of great importance for scientific interest.Nguyen-Trung3and McGlynn4et al.have made experimental studies on the frequencies of O=U=O vibrations in uranyl ion with various inorganic and organic ligands.de Jong et al.5have theoretically reported the properties of geometries and frequencies on monomer complexes(n=1-3)and trimer complexesusing local density functional theory in gaseous phase with the program of NWChem.Our previous study6by Gaussian 03 program established linear correlations between the frequency of O=U=O systematical vibration and the number of different ligands in gaseous and aqueous phases. And some other spectroscopic techniques,such as X-ray diffraction,nuclear magnetic resonance spectroscopy(NMR),and extended X-ray absorption fine structure(EXAFS),have been employed for uranyl carbonate studies.7,8Some reviews have summarized recent advances in computational actinide chemistry.9-12UV-Vis spectra of uranyl(VI)carbonate complexes have been obtained in experiments,13,14but few studies were reported from the theoretical perspective.Su et al.9,15have calculated the luminescence properties of uranyl-glycine-water complexes in solution with the statistically averaged orbital potentials (SAOP)employed in the spin-orbit coupling time-dependent density functional theory(SO-TD-DFT)calculations.And for other actinide elements,the spectrum studies in theory and experiment were also limited.16To explain the absorption or emission spectra of actinide complexes is important for environmental detections.
As seen from above survey,there is much experimental and theoretical interest in uranyl carbonate complexes.Absorption spectroscopy is a very important tool for analyzing chemical systems,but the interpretation of electronic spectra in terms of molecular structures remains challenges for experiments.In this paper,solvent effects on geometries of non-hydrated and hydrated structures of uranyl carbonate complexes were considered to predict characteristic absorption band of uranyl complexes in aqueous phase.
2 Calculation details
It is necessary to include relativistic effects on uranium included system.There are two general classes of relativistic effects that are clearly summarized as scalar relativistic and spinorbit coupling relativistic by Kaltsoyannis et al.17The scalar relativistic method is sufficient for ground stateʹs properties of actinide systems,including molecular geometries and vibration frequencies.5,15,17However,the spin-orbit coupling relativistic methods are demanded to be included in the calculation of excited states properties especially optical excitation energies.
It is advised to obtain results by using gradient-correction DFT functional with small-core relativistic effective core potential(RECP)in the benchmark for approximate calculations on bare uranyl ion by de Jong et al.18We performed the optimizations for ground state structures on the Amsterdam Density Functional Code(ADF2010.01).19-21The corresponding approximation methods obtained reliable results on uranyl glycine complexes by Su et al.15Hence,we used the Perdew-Burke-Ernzerhof(PBE)exchange-correlation functional22and uncontracted Slater basis sets of triple-ζ plus one polarization(TZP) quality for the U atom and sets of double-ζ plus one polarization(DZP)quality for the C and O atoms from ADF basis library.23Small frozen atomic core approximation was applied to C,O with[1s2]and to U with[1s2-5d10].The zeroth-order regular approximation(ZORA)was used to account for scalar relativistic effects.24-26Solvent effects which were estimated by the conductor-like screening model(COSMO)used the solvent accessible surface(SAS).27,28And the default water dielectric constant(ε)is 78.4 for aqueous phase calculations.
Electronic transitions by the time-dependent density functional theory(TD-DFT)29calculations based on ground state of uranyl complexes were performed.We examined the transitions from the ground state to the excited states.And the spinorbit coupling was included in excitation energies by the relativistic two-component ZORA formalism.24-26The statistically averaged orbital potentials(SAOP)15,30were employed in the TD-DFT calculations.
3 Results and discussion
3.1 Bond distance
Geometries of uranyl carbonate complexes have become great interesting for theoretical and experimental chemists. From previous theoretical studies,5,31the calculated bond lengths for uranyl tri-carbonate anions are in accordance with experimental results8,32(Table 1).The use of COSMO improves the value of R(U-Ocarbonate)in structures at the PBE and PW91 levels.Although addition of diffused functions to U at the LDA
3level leads to decrease of U=O bond length,the method is difficult in converging.5The R(U-Ocarbonate)at the PBE/ZORA level is close to experimental data in aqueous phase8as compared to those with the PW91 and LDA methods.Hence,we use the PBE/ZORA method with the the approximations of successive uranyl carbonate complexes.
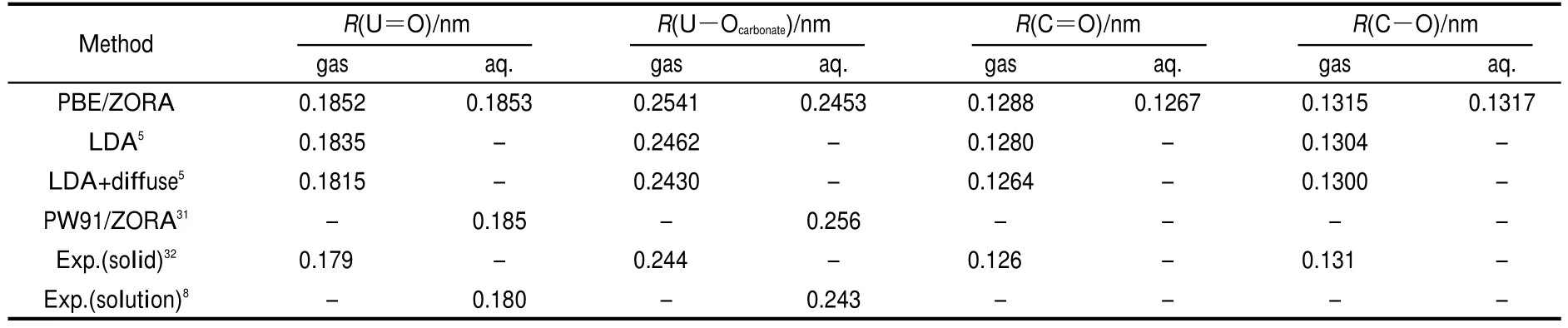
Table 1 Comparison of calculated and experimental bond lengths for UO2(CO3)4-
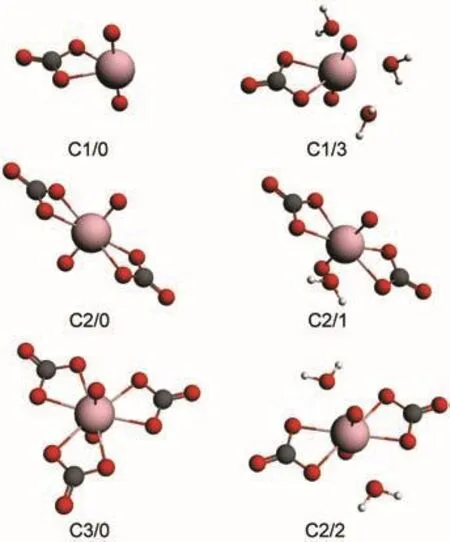
Fig.1 Stable structures of non-hydrated(C1/0,C2/0,C3/0)and hydrated(C1/3,C2/1,C2/2)uranyl carbonate complexes
The coordination number(CN)in the equator of the linear UO2unit for stable uranyl complexes is 5.15,33,34When CNs of uranyl carbonate complexes are 2 or 4,water molecule could be added to the equator to form more stable structures.Here we label the non-hydrated form and hydrated form with Cn/m (n and m are the numbers of carbonate and water ligands in uranyl complexes,respectively).The non-hydrated(C1/0,C2/0, C3/0)and hydrated(C1/3,C2/1,C2/2)structures of uranyl complexes are shown in Fig.1.In order to estimate the ligand effects on geometry and electronic structures of uranyl complexes,PBE/ZORA method is employed to optimize both forms.The bond lengths are listed in Table 2.
The trend in the bond distances of non-hydrated structures is substantially the same as those observed in experimental and theoretical data.5By adding carbonate ligand in the equator of the linear UO2unit,the U=O and U-Oeqare elongated.It seems that ligands competing effects of carbonate ions have weakened the bonds of U=O and uranium-carbonate coordination.The R(U=O)of the successive uranyl carbonate complexes ranges from 0.1798 to 0.1852 nm in gaseous phase and 0.1802 to 0.1853 nm in aqueous phase.R(U=O)of C1/0 (UO2CO3)is widely discussed in experiments.35-38It arrived at 0.193 nm by Christ et al.,35(0.167±0.009)nm by Cromer and Harper,36and 0.174 nm by Finch et al.37Recently,Matar38has carried out a DFT study on lattice anisotropy,electronic and chemical geometry of C1/0,and the calculated R(U=O)value is 0.179 nm in the uranyl carbonate crystal.And we favor the results of longer U=O of C1/0 as those determined by theoretical methods.Solvent effects of water elongate the U=O in C1/ 0 as compared with that in gaseous phase.But for high coordination complexes of C2/0 and C3/0(uranyl di-,tri-carbonate complexes),similar R(U=O)values are obtained in gaseous and aqueous phases.The carbonate ion performs a great geometry deformation in successive uranyl carbonate complexes. The differences between R(C=O)and R(C-O)are great in the complexes which contain one or two uranium-carbonate coordination bonds,but are tiny in the complex with three uranium-carbonate coordination bonds.The hydrated water elongates U=O of C1/3 as compared with that in C1/0.And in C2/ 0,C2/1,and C2/2,similar bond lengths in gaseous and aqueous phases again illustrate the little effect by solvent on high uranium-carbonate coordination complexes.The fact that UOwateris much longer than U-Ocarbonateindicates that hydrated water may interact with uranyl ion weakly.
3.2 Binding energy
The corresponding binding energies15of the Cn/m complexes defined by energy differences between the whole complex and its components are based on the following formula:
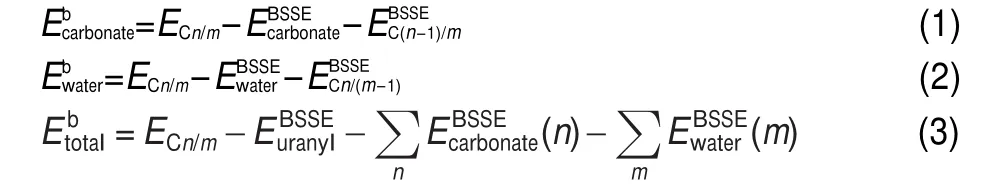
Cn/m refers to the non-hydrated and hydrated structures of ura-nyl complexes.And C(n-1)/m or Cn/(m-1)refers to the structures removing one carbonate ion or one water molecule from Cn/m.Carbonate,water,and uranyl are the components of a whole complex.The E refers to energy with the inclusion of spin orbit coupling relativistic effects in gaseous or aqueous phase.And all componentsʹenergies for the whole complex include the basis set superposition error(BSSE)correction.
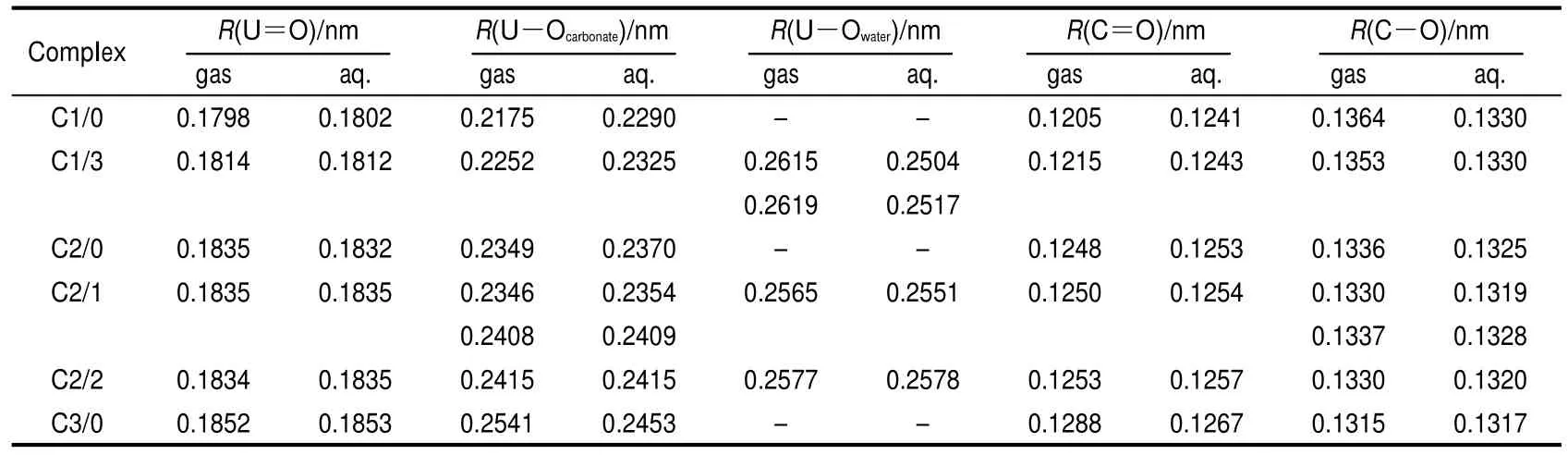
Table 2 Bond distances for uranyl carbonates in gaseous and aqueous phases
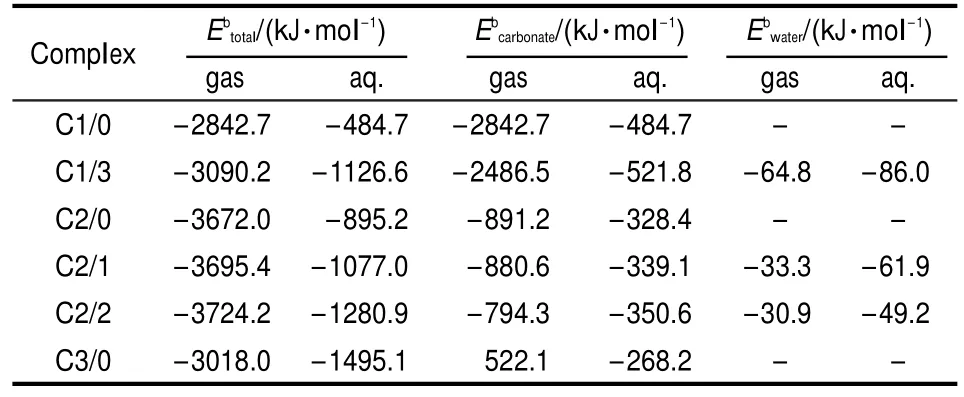
Table 3 Binding energies for uranyl carbonates in gaseous and aqueous phase
The parameter Ebcarbonate(Table 3)introduced by adding hydrated water in structures indicates that the bond of uraniumcarbonate coordination is strengthened in uranyl hydrated structures.And the small value of Ebwaterillustrates that uranium-water coordination is far weaker.In aqueous phase,the total binding energyof C3/0 is-1495.1 kJ·mol-1and the negative value means that the formation of C3/0is exothermic.The total binding energies show that C2/2 and C3/0 are the most stable carbonate complexes in gaseous and aqueous phases,respectively. These results agree with stability constants of uranyl carbonate in experiments.13,14,39In gaseous phase,the binding energy ofwith positive value means the formation of C3/0 from reactions between carbonate ion and C2/0 is endothermic by 522.1 kJ·mol-1.In Table 3,although hydrated water seems to increase the total binding energies of uranyl carbonate complexes,ligands competing effects could decrease the energies to separate one carbonate ion from C1/3,C2/1,and C2/2.The bond of uranium-water coordination tends to be intensified in aqueous phase as the binding energies of U-Owaterare higher than those in gaseous phase(Table 3).
3.3 Molecular orbital
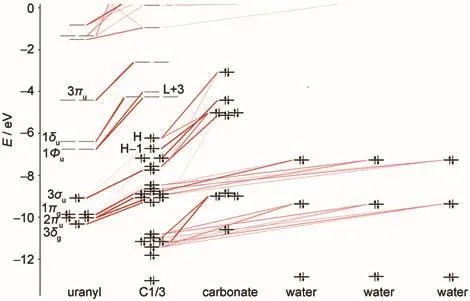
Fig.2 Molecular orbital diagrams of hydrated uranyl complex C1/3H and L are short for the highest occupied and lowest unoccupied orbitals, respectively.The same abbreviation scheme is applied from Fig.3 to Fig.6.
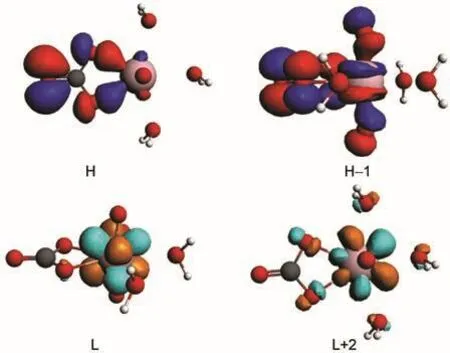
Fig.3 Isosurfaces(Ψ=±0.03 a.u.)of the corresponding frontier orbitals(H,H-1,L,L+2)in C1/3 complex
Molecular orbital(MO)diagrams and frontier orbitals of C1/ 3 are displayed in Fig.2 and Fig.3,respectively.In uranyl,the highest occupied MOs(HOMOs)3σg,3σu,1πg,and 2πuare most components of 2p shell centered on oxygen atoms,while the lowest unoccupied MOs(LUMOs)1Фu,1δu,and 3πualmost are the components of 5f shell centered on uranium atom.The HOMOs of C1/3 consist of 2p components from carbonate ligand and the LUMOs are mainly uranium 5f unoccupied orbital as the fragments interactions illustrated in Fig.2.Here we find that the H-1 orbital comes from the interactions of 3σuorbital of uranyl and 2p orbital of carbonate.And the HOMOs, 3σg,1πg,and 2πuof uranyl come to interact with 2p components of water ligands to form orbital of uranyl complex in low energy level.The corresponding orbitals shown in Fig.3 are in accordance to the molecular orbitals interactions.Hence,the more carbonate ligands come to ligate with uranyl ion,the more 2p components of carbonate based orbital could insert in the high occupied energy level.That is also true in molecular orbital diagrams of C2/2 and C3/0 shown in Fig.4 and Fig.5. However,the main difference compared to C1/3 complex is that the components of 5f shell begin to take part in bonding interactions in C2/2 and C3/0.The bonding orbital between 5f and 2p components of C2/2 and C3/0 are showed in Fig.6.And we may attribute stability of C3/0 carbonate complex in aqueous phase to the inclusion of 5f components in high occupied orbital.
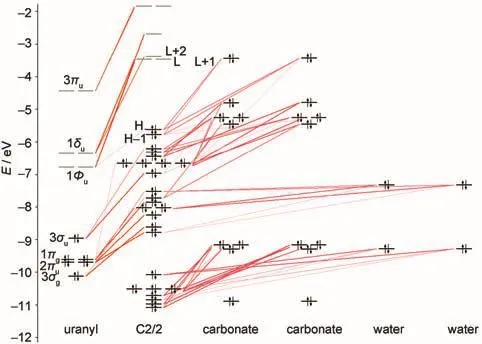
Fig.4 Molecularorbitaldiagramsof hydrated uranylcomplexC2/2
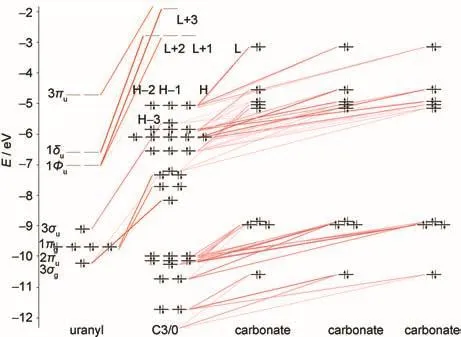
Fig.5 Molecular orbital diagrams of hydrated uranyl complex C3/0
3.4 Electronic transitions
Electronic transition calculations from PBE-ZORA by ADF program are mentioned in the detail of calculations.State splitting by the ligand field and spin-orbit coupling makes more transitions available.But symmetry in structure would restrict the transitions as selection rule only allows transitions with parity changes.
The experimental characteristic absorption band was at 414 nm for uranyl species.40The absorption band of the uranyl complexes was broad in the visible range between 520 and 370 nm by extensive spectroscopy experiments studies on uranyl carbonate complexes13,14,41,42and some other inorganic ligands.38,43-47The excitation energies above 370 nm of uranyl carbonate complexes in Table 4 are in accordance with the major characteristics observed from experiments discussed above. Just as the main assignments of excitations transitions shown in Table 4,transitions are essentially transferred from high occupied orbital to the low vacant orbital.As the molecular orbitals discussed above,the main transitions between HOMOs and LUMOs are clearly ligand-to-metal charge transfer(LMCT) with electron density essentially transferring from carbonates ligand-based 2p components towards the vacant 5f orbital of the uranium.Only a few high occupied orbitals come from the interactions of 3σuorbital of uranyl and 2p components based orbital of carbonate.When compared to the allowed transitions in C1/3,the allowed ones in C2/2 and C3/0 turn to be with a smaller number for the restriction of centro symmetry of molecule in the selection rule.The maximum of wavelengths are blue shifted for uranyl carbonate complexes C1/3 to C3/0.And relatively stronger absorptions at 420 nm in all the uranyl carbonate complexes accord with properties of material itself.But our calculations indicate that absorptions at 420 nm in C2/2 and C3/0 are far stronger than those in C1/3.Our calculated UV-Vis absorption spectra are simulated by SO-TD-DFT calculations and displayed in Fig.7.The characters of uranyl carbonate complexes on UV-Vis spectra depend on the coordinating number of carbonate ion.And the additions of ligated water in structures make UV-Vis spectra slightly red shift as compared to those in the corresponding non-hydrated structures.Uranyl carbonate C1/0 behaved differently in absorption spectrum with no major characteristics as compared to those of other uranyl carbonate species by experimental study.44And the low intensity of absorptions for C1/3 is in accordance to the results from experiments.44The UV-Vis absorption spectra observed by Gong et al.47on uranyl acetohydroxamate also show no characteristics of other uranyl complexes.The absorption intensity of uranyl carbonate complexes can be explained by the differences in molecular orbital.As we have discussed above, components of 5f shell begin to take part in bonding orbital interactions in C2/2 and C3/0.The ligand contributions of carbonate ion allow larger ligand-to-metal charge transfer(LMCT). We can find that the carbonate ligand plays an important role in electronic absorption.It indicates that the addition of carbonate ligand leads to a blue shift in the maximum wavelength and high intensity of absorption in the near visible region.
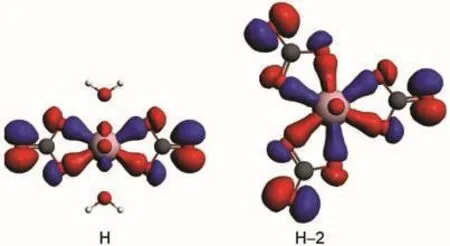
Fig.6 Isosurfaces(Ψ=±0.03 a.u.)of the bonding orbitals(H,and H-2)in C2/2 and C3/0 complexes
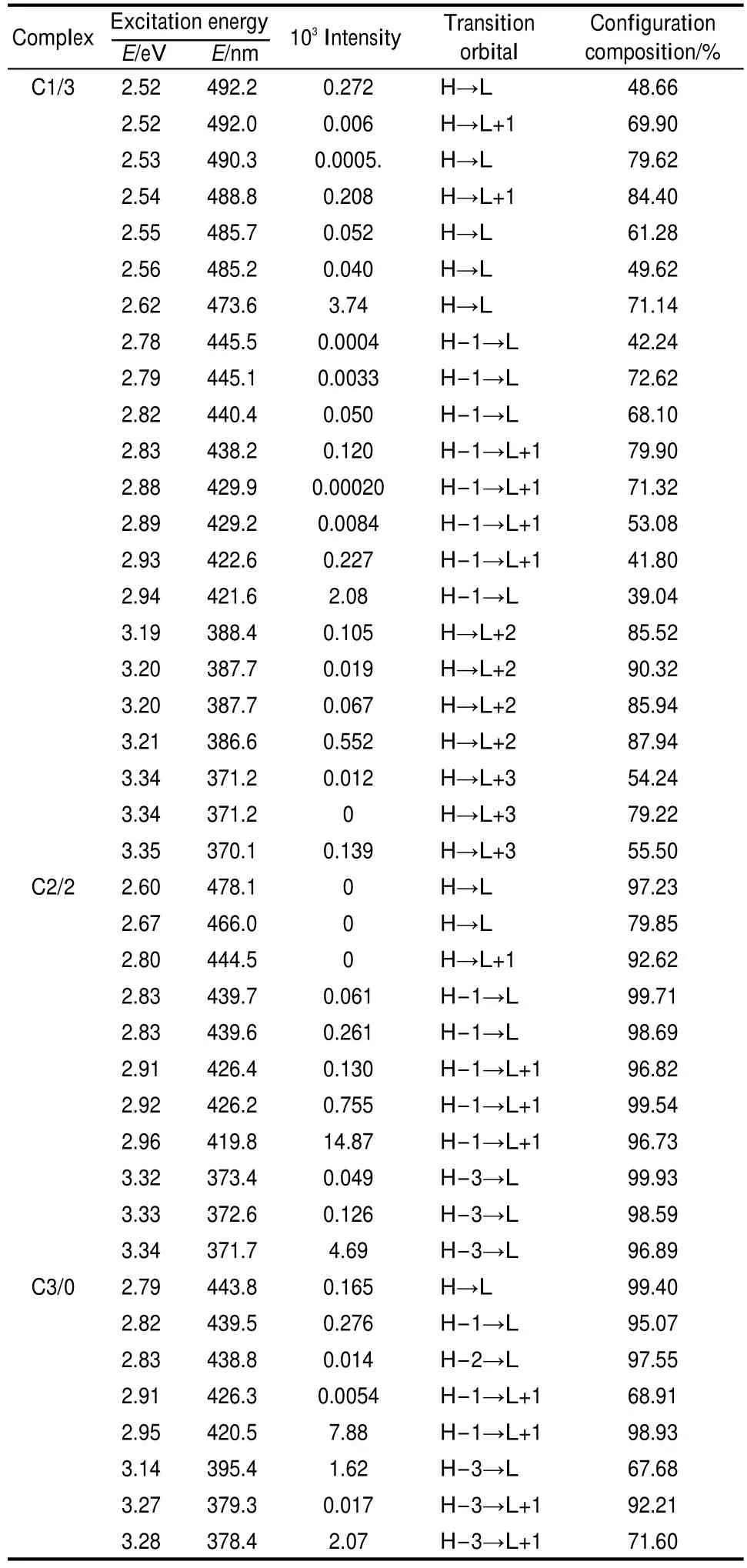
Table 4 Excitation energy and main assignments of spinpolarized excitations for uranyl carbonate complexes
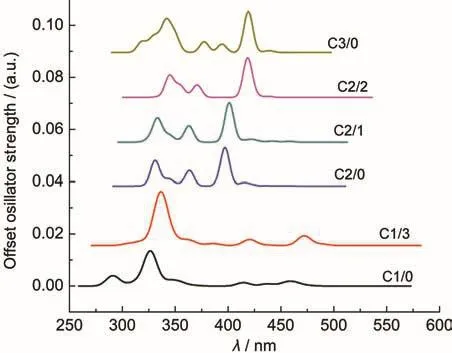
Fig.7 Simulated UV-Vis spectra for non-hydrated(C1/0,C2/0, C3/0)and hydrated(C1/3,C2/1,C2/2)uranyl carbonate complexes as carbonate and hydrated water ligands changed from 1 to 3
4 Conclusions
It indicates that carbonate ligand plays an important role in its geometrical and electronic transitional properties.The geometries of uranyl carbonate complexes agree well with the available experimental and theoretical studies.The interpretations of the characteristics of uranyl carbonate complexes were suggested from a certain viewpoint of molecular orbital.The differences among orbital compositions show that more 2p based orbitals from carbonate ligands in the high occupied energy level are due to the addition of carbonate ligands in structures.This promotes the LMCT in the near visible region.And the adding of water in uranyl complexes makes little effect on LMCT transitions.Hence,the low intensity of absorptions at near visible region for C1/3 is attributed to the low number of carbonate ligand.
(1)Clark,D.L.;Hobart,D.E.;Neu,M.P.Chem.Rev.1995,95,25.
(2) Meinrath,G.J.Radioanal.Nucl.Chem.1996,211,349.
(3)Nguyen-Trung,C.;Begun,G.M.;Palmer,D.A.Inorg.Chem. 1992,31,5280.
(4) McGlynn,S.P.;Smith,J.K.;Neely,W.C.J.Chem.Phys.1961, 35,105.
(5) de Jong,W.A.;Aprà,E.;Windus,T.L.;Nichols,J.A.;Harrison, R.J.;Gutowski,K.E.;Dixon,D.A.J.Phys.Chem.A 2005, 109,11568.
(6) Gu,J.F.;Lu,C.H.;Chen,W.K.;Xu,Y.;Zheng,J.D.Acta Phys.-Chim.Sin.2009,25,655. [辜家芳,陆春海,陈文凯,许 莹,郑金德.物理化学学报,2009,25,655.]
(7)Allen,P.G.;Bucher,J.J.;Clark,D.L.;Edelstein,N.M.; Ekberg,S.A.;Gohdes,J.W.;Hudson,E.A.;Kaltsoyannis,N.; Lukens,W.W.Inorg.Chem.1995,34,4797.
(8) Docrat,T.I.;Mosselmans,J.F.W.;Charnock,J.M.;Whiteley, M.W.;Collison,D.;Livens,F.R.;Jones,C.;Edmiston,M.J. Inorg.Chem.1999,38,1879.
(9) Su,J.;Li,J.Prog.Chem.2011,23,1329.[苏 静,李 隽.化学进展,2011,23,1329.]
(10) Wang,D.Q.;Gunsteren,W.F.v.Prog.Chem.2011,23,1566. [王东琪,Gunsteren,W.F.v.化学进展,2011,23,1566.]
(11) Hu,H.S.;Wu,G.S.;Li,J.J.Nucl.Radiochem.2009,31,25. [胡憾石,吴国是,李 隽.核化学与放射化学,2009,31,25.]
(12) Liu,W.J.Prog.Chem.2007,19,833.[刘文剑.化学进展, 2007,19,833.]
(13) Cinnéide,S.Ó.;Scanlan,J.P.;Hynes,M.J.J.Inorg.Nucl. Chem.1975,37,1013.
(14) Scanlan,J.P.J.Inorg.Nucl.Chem.1977,39,635.
(15) Su,J.;Zhang,K.;Schwarz,W.H.E.;Li,J.Inorg.Chem.2011, 50,2082.
(16) Matsika,S.;Pitzer,R.M.;Reed,D.T.J.Phys.Chem.A 2000, 104,11983.
(17) Kaltsoyannis,N.;Hay,P.J.;Li,J.;Blaudeau,J.P.;Bursten,B. E.Theoretical Studies of the Electronic Structure of Compounds of theActinide Elements.In The Chemistry of the Actinide and Transactinide Elements;Morss,L.R.,Edelstein, N.M.,Fuger,J.,Eds.;Springer:Netherlands,2006;p 1893.
(18) de Jong,W.A.;Harrison,R.J.;Nichols,J.A.;Dixon,D.A. Theor.Chem.Acc.2001,107,22.
(19)ADF2010,SCM,Theoretical Chemistry;Vrije Universiteit: Amsterdam,The Netherlands;http://www.scm.com.
(20) Guerra,C.F.;Snijders,J.G.;Velde,G.T.;Baerends,E.J.Theor. Chem.Acc.1998,99,391.
(21) Velde,G.T.;Bickelhaupt,F.M.;Baerends,E.J.;Guerra,C.F.; van Gisbergen,S.J.A.;Snijders,J.G.;Ziegler,T.J.Comput. Chem.2001,22,931.
(22) Perdew,J.P.;Burke,K.;Ernzerhof,M.Phys.Rev.Lett.1996, 77,3865.
(23) van Lenthe,E.;Baerends,E.J.J.Comput.Chem.2003,24, 1142.
(24) van Lenthe,E.;Ehlers,A.E.;Baerends,E.J.J.Chem.Phys. 1999,110,8943.
(25) van Lenthe,E.;Baerends,E.J.;Snijders,J.G.J.Chem.Phys. 1994,101,9783.
(26) van Lenthe,E.;Baerends,E.J.;Snijders,J.G.J.Chem.Phys. 1993,99,4597.
(27) Lee,B.;Richards,F.M.J.Mol.Biol.1971,55,379.
(28) Richards,F.M.Annu.Rev.Biophys.Bioeng.1977,6,151.
(29) Perdew,J.P.;Ruzsinsky,A.;Tao,J.;Staroverov,V.N.;Scuseria, G.E.;Csonka,G.I.J.Chem.Phys.2005,123,062201.
(30) Schipper,P.R.T.;Gritsenko,O.V.;van Gisbergen,S.J.A.; Baerends,E.J.J.Chem.Phys.2000,112,1344.
(31) Vázquez,J.;Bo,C.;Poblet,J.M.;de Pablo,J.;Bruno,J.Inorg. Chem.2003,42,6136.
(32) Graziani,R.;Bombieri,G.;Forsellini,E.J.Chem.Soc.Dalton Trans.1972,2059.
(33) Spencer,S.;Gagliardi,L.;Handy,N.C.;Ioannou,A.G.; Skylaris,C.K.;Willetts,A.;Simper,A.M.J.Phys.Chem.A 1999,103,1831.
(34) Bardin,N.;Rubini,P.;Madic,C.Radiochim.Acta 1998,83,189.
(35) Christ,C.L.;Clark,J.R.;Evans,H.T.J.Science 1955,121, 472.
(36) Cromer,D.T.;Harper,P.E.Acta Crystallogr.1955,8,847.
(37)Finch,R.J.;Cooper,M.A.;Hawthorne,F.C.;Ewing,R.C. Can.Mineral.1999,37,929.
(38) Matar,S.F.Chem.Phys.2010,372,46.
(39) Pashalidis,I.;Czerwinski,K.R.;Fanghanel,T.;Kim,J.I. Radiochim.Acta 1997,76,55.
(40) Rude,W.Los Alamos Science 2000,26,412.
(41) Meinrath,G.J.Radioanal.Nucl.Chem.1997,224,119.
(42) Meinrath,G.;Klenze,R.;Kim,J.I.Radiochim.Acta 1996,74, 81.
(43) Havel,J.;Soto-Guerrero,J.;Lubal,P.Polyhedron 2002,21, 1411.
(44) Tian,G.;Rao,L.J.Chem.Thermodyn.2009,41,569.
(45) Rao,L.;Tian,G.J.Chem.Thermodyn.2008,40,1001.
(46) Tian,G.;Rao,L.Inorg.Chem.2009,48,6748.
(47) Gong,C.M.S.;Poineau,F.;Czerwinski,K.R.Radiochim.Acta 2007,95,439.
September 26,2011;Revised:January 8,2012;Published on Web:January 17,2012.
Electronic Structures of Uranyl(VI)Carbonate Complexes in the Aqueous Phase
GU Jia-Fang1LU Chun-Hai2CHEN Wen-Kai1,*CHEN Yong1XU Ke1HUANG Xin1ZHANG Yong-Fan1
(1Department of Chemistry,Fuzhou University,Fuzhou 350108,P.R.China;2College of Nuclear Technology and Automation Engineering,Chengdu University of Technology,Chengdu 610059,P.R.China)
A systematic study of series of non-hydrated and hydrated Cn/m uranyl carbonate complexes (n is number of carbonate ligands,and m is number of water molecules)in the aqueous phase was carried out using relativistic density functional theory.The conductor-like screening model was used to calculate solvent effects.The zeroth-order regular approximation was used to account for scalar relativistic effects and spin-orbit coupling relativistic effects.Time-dependent density functional theory with the inclusion of spin-orbit coupling relativistic effects was used to calculate electronic transitions using the statistically averaged orbital potentials.The results indicate that carbonate ligands play an important role in the geometric and electronic transition properties of the complex.The stability of the C3/0 carbonate complex in the aqueous phase may be attributed to the involvement of 5f components in the highest occupied bonding orbital.The addition of carbonate ligands caused a blue shift in the maximum wavelength and high intensity absorptions in the near visible region.
Uranyl;UV-Vis;Solvent effect;Time-dependent density functional theory;Spin-orbit coupling relativistic effect
10.3866/PKU.WHXB201201171
O641
∗Corresponding author.Email:qc2008@fzu.edu.cn;Tel:+86-591-22866162.
The project was supported by the National Natural Science Foundation of China(10676007)and Program for New Century Excellent Talents at the University of Fujian Province,China(HX2006-103).
国家自然科学基金(10676007)和福建省高等学校新世纪优秀人才计划(HX2006-103)资助项目
猜你喜欢
科学大众(2023年17期)2023-10-26 07:38:56
小天使·二年级语数英综合(2021年5期)2021-07-11 10:58:35
化学与生物工程(2021年2期)2021-02-25 12:56:46
无机材料学报(2020年2期)2020-03-08 14:05:54
分析化学(2019年3期)2019-03-30 10:59:24
中学化学(2017年2期)2017-04-01 08:51:54
材料科学与工程学报(2016年4期)2017-01-15 13:35:48
合成化学(2015年4期)2016-01-17 09:01:11
核科学与工程(2015年2期)2015-09-26 11:57:21
无机化学学报(2014年7期)2014-02-28 17:32:10
