
分子靶向药物PI3K抑制剂的研究进展
2012-11-26 12:38金美花孔德新
药品评价 2012年5期
金美花,孔德新
天津医科大学药学院 天津市临床药物关键技术重点实验室、基础医学研究中心,天津 300070
20世纪末期到21世纪初期分子靶向药物的出现是抗癌药物研究进展的里程碑式的事件。分子靶向药物是指作用于肿瘤细胞的特定分子靶点,对肿瘤细胞的增殖、侵袭、转移等恶性生物学行为具有抑制作用从而产生抗肿瘤作用的药物。这些分子靶点在肿瘤组织中特异性表达或者表达较高,而在正常组织不表达或表达较少。因此分子靶向药物一般能够选择性地杀伤肿瘤细胞或抑制其生长增殖,而对正常细胞的毒性作用较小。
磷脂酰肌醇3-激酶 (phosphatidylinositol 3-kinase, PI3K) 为一种由调节亚单位p85或p101和催化亚单位p110组成的脂激酶,通过催化磷脂酰肌醇4,5-二磷酸(phosphatidylinositol 4,5-bisphosphate,PIP2)磷酸化为磷脂酰肌醇3,4,5-三磷酸(phosphatidylinositol 3,4,5-trisphosphate, PIP3)而激活下游的Akt等从而对细胞的增殖、生存和代谢等起关键作用。由于PI3K与癌症等疾病的密切关系,以PI3K为靶标的抑制剂的开发引起了国际制药界的高度重视。本文首先简单介绍PI3K的功能和分类,然后就PI3K抑制剂的研究进展做一综述。
1 PI3K的功能及与各种疾病的关系
PI3K是一个脂激酶家族,它们可催化PIP2磷酸化为PIP3而激活下游的Akt[1]。与PI3K的功能相反, 肿瘤抑制基因PTEN(phosphatase and tension homolog deleted on chromosome ten)使PIP3去磷酸化生成PIP2(图1)。PI3K基因突变和扩增在癌症中频繁发生[2]以及PTEN在癌症中缺失等都提示PI3K与肿瘤发生的密切关系[3]。
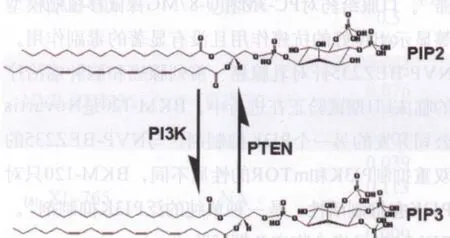
图1 PI3K和PTEN的功能示意图Fig 1 Schematic representation of the functions of PI3K and PTEN
PI3K依其结构和磷酸化底物的不同被分为Ⅰ、Ⅱ、Ⅲ三种类型。I型PI3K是由一个调节亚单位p85和一个催化亚单位p110组成的二聚体,一般催化PIP2磷酸化而生成PIP3。Ⅱ型PI3K包含三个成员,分别为PI3KC2α、PI3KC2β和PI3K2ɣ,它们一般磷酸化磷脂酰肌醇(phosphatidylinositol, PI)和磷脂酰肌醇4-磷酸(phosphatidylinositol 4-phosphate,PIP),其功能主要与膜运输等有关。Ⅲ型PI3K只有一个成员Vps34(vacuolar protein sorting 34),它磷酸化PI生成磷脂酰肌醇3-磷酸。Vps34关于胞吞作用和囊泡运输的功能已广为人知。近年来又有报道关于Vps34与自噬的关系:它对于自噬现象的发生是不可或缺的。由于迄今为止功能研究最多的是I型PI3K,因此人们通常所说的PI3K就是I型PI3K。另外,雷帕霉素靶体蛋白(mammalian target of rapamycin, mTOR)和DNA依赖性蛋白激酶(DNA-dependent protein kinase, DNA-PK)等蛋白激酶结构与p110相似,所以有时被分类为IV型PI3K。I型PI3K依它们的调节亚单位和上游调节分子的不同又分为IA和IB两个亚类。IA亚类PI3K可被各种受体酪氨酸激酶(receptor tyrosine kinase, RTK)和Ras激活, 包括PI3Kα、PI3Kβ和PI3Kδ三个亚型,分别由各自的催化亚单位p110与调节亚单位p85组成。IB亚类PI3Kɣ由催化亚基p110ɣ和调节亚基p101或p84组成,它主要被G蛋白耦联受体(G-protein-coupled receptor, GPCR)激活。关于各种亚型PI3K的体内分布,PI3Kα和PI3Kβ在各种器官均有表达,而PI3Kδ和PI3Kɣ主要分布在骨髓细胞中。四种亚型的功能也有所不同,由于被报道的癌症患者中PI3K基因的变异均为编码p110α的基因PIK3CA的变异,因而PI3Kα被认为在肿瘤发生中起着重要的作用。另外PI3Kα还与胰岛素信号传导和葡萄糖代谢有关。PI3Kβ亚型可激活血小板,因此在血栓性疾病的发展中起着重要的作用;此外,PI3Kβ近年来被报道在PTEN缺失的癌症患者中起着比PI3Kα亚型还重要的作用。PI3Kδ和PI3Kɣ与炎症和免疫等有密切的关系[4,5]。
PI3K作为治疗肿瘤的靶点引人注目,是因为它位于众多重要信号通路的关键性的信号位置[4,5](图2)。PI3K被RTK或Ras激活后催化PIP2生成PIP3。PIP3与Akt和3-磷酸肌醇依赖性蛋白激酶(3-phosphoinositide dependent protein kinase, PDK)等蛋白激酶结合,激活并将其募集到细胞膜。Akt除了直接被PIP3激活以外,还会被PDK和mTORC2(mTOR complex 2)所激活。Akt可通过抑制GSK3来稳定细胞周期素D1(cyclin D1)的表达以及抑制FOXO调节的周期素依赖性蛋白激酶(cyclin dependent kinase, Cdk)抑制剂p27的转录而促进细胞周期的进行;Akt还通过抑制BAD (Bcl2-antagonist of cell death) 维持细胞生存;又可磷酸化mTORC1来促进蛋白质的合成和细胞生长[6]; 还可通过mTORC1上调缺氧诱导因子1α(hypoxia-inducible factor 1α,HIF-1α和血管内皮生长因子(vascular endothelial growth factor, VEGF)而促进血管生成[5]。但是mTORC1/S6K通路又负性调控胰岛素受体底物(insulin receptor substrate, IRS)[5],所以mTORC1的阻断会激活上游蛋白如PI3K和Akt,从而减弱它们的抑制能力,这也是单纯抑制mTOR靶点的缺点[5]。
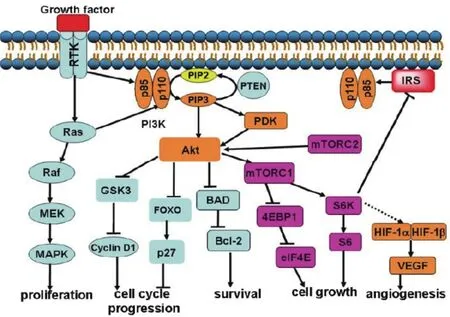
图2 PI3K/Akt信号通路在癌症发生中的作用Fig 2 The PI3K/Akt pathway in tumorigenesis.
如上所述,PI3K在肿瘤发生中起着非常重要的作用,因此PI3K抑制剂作为抗肿瘤药物的开发引起了制药界的高度重视。
2 PI3K抑制剂的研究进展
LY294002 和Wortmannin是两种经典的PI3K抑制剂,它们虽显示了一定的体内和体外抗肿瘤作用,但由于分别引起皮肤和肝脏毒性以及溶解度与稳定性等原因, 未能进入临床试验[5]。
2004年一篇发表在《Science》上关于PIK3CA变异在各种癌症中被发现的报道[2]引起了广大制药界科研工作者的兴趣,也打响了PI3K抑制剂开发竞赛的发令枪。PI3K晶体结构的阐明更加速了PI3K抑制剂的药物设计和发现过程。此后,专一性更强的新型PI3K抑制剂不断出现。这些抑制剂大体可分为PI3K亚型特异性抑制剂、泛PI3K抑制剂和PI3K/mTOR双重抑制剂,见表1。
CAL-101(IC87114)是Calistoga公司开发的一种PI3Kδ特异性抑制剂, 其IC50为0.5 μM, 是PI3Kɣ IC50的58倍,PI3Kα和PI3Kβ IC50的100多倍[5]。由于PI3Kδ在炎症中的重要作用,最初关于CAL-101的药理研究集中在其抗炎和治疗自身免疫性疾病上。CAL-101显示出了对炎症和小鼠哮喘模型的治疗效果。由于PI3Kδ主要在骨髓细胞中表达,CAL-101用于治疗白血病的研究近几年来受到重视。研究表明CAL-101可抑制急性骨髓性白血病细胞的增殖,同时并不影响正常造血细胞的增殖。此外,与mTOR抑制剂RAD001或拓扑异构酶II抑制剂VP16联合用药,能够显著提高它们抑制白血病细胞增殖的效果[5]。CAL-101已进入治疗白血病的临床Ⅱ期试验。
NVP-BEZ235是Novartis公司开发的一种PI3K/mTOR双重抑制剂。它抑制PI3Kα、β、δ和ɣ的IC50分别为4、76、5和7nM[7]。在体外,NVP-BEZ235强力地抑制肿瘤细胞的增殖,且诱导G1期细胞周期阻滞[7]。口服给药对PC-3M和U-87MG裸鼠移植瘤模型等显示出良好的抗癌作用且没有显著的毒副作用。NVP-BEZ235针对乳腺癌、前列腺癌和脑肿瘤治疗的临床I/II期试验正在进行中。BKM-120是Novartis公司开发的另一个PI3K抑制剂,与NVP-BEZ235的双重抑制PI3K和mTOR的性质不同,BKM-120只对PI3K有抑制活性,是一种单纯的泛PI3K抑制剂[8]。BKM-120已进入临床Ⅱ期试验。
GDC-0941是Genentech公司开发的泛PI3K抑制剂。其对PI3Kα、β、δ和ɣ的IC50分别为3、33、3和75nM,但对mTOR无明显抑制作用[9]。口服用药对多种裸鼠移植瘤模型显示良好的抗癌作用。而且与多西他赛等联合用药,可明显增强其疗效。GDC-0980是该公司开发的另一种PI3K抑制剂,它与GDC-0941的区别在于对mTOR也有抑制作用,是一种PI3K/mTOR双重抑制剂[8]。GDC-0941和GDC-0980均已进入临床I期试验。
ZSTK474是由Zenyaku Kogyo公司和日本癌研究会合作开发的一种泛PI3K抑制剂。ZSTK474对PI3Kα、β、δ和ɣ的IC50分别为16、44、5和49nM[10]。在体外ZSTK474可抑制39种人癌细胞系的细胞增殖,其GI50(达到50%增殖抑制所需的药物浓度)为0.32 μM[11]。与多数其它PI3K抑制剂类似,可诱导各种肿瘤细胞的细胞周期停滞在G1期,且没有出现明显的细胞凋亡。其阻滞细胞周期的机理可能归因于cyclinD1的失活、p27表达增加和pRB的去磷酸化作用[8]。有研究者曾系统研究了ZSTK474的体内和体外抗血管生成作用[12]。ZSTK474 可抑制肾癌细胞RXF-631L中HIF-1α的表达和VEGF的分泌,而且抑制血管内皮细胞的增殖、迁移和体外小管形成(图4A)。在体内试验中显著抑制了RXF-631L裸鼠移植瘤小鼠肿瘤组织中的毛细血管数。ZSTK474口服用药对多种裸鼠移植瘤模型显示出良好的的抗肿瘤效果(图4B),现正在进行临床I期试验评价[8]。
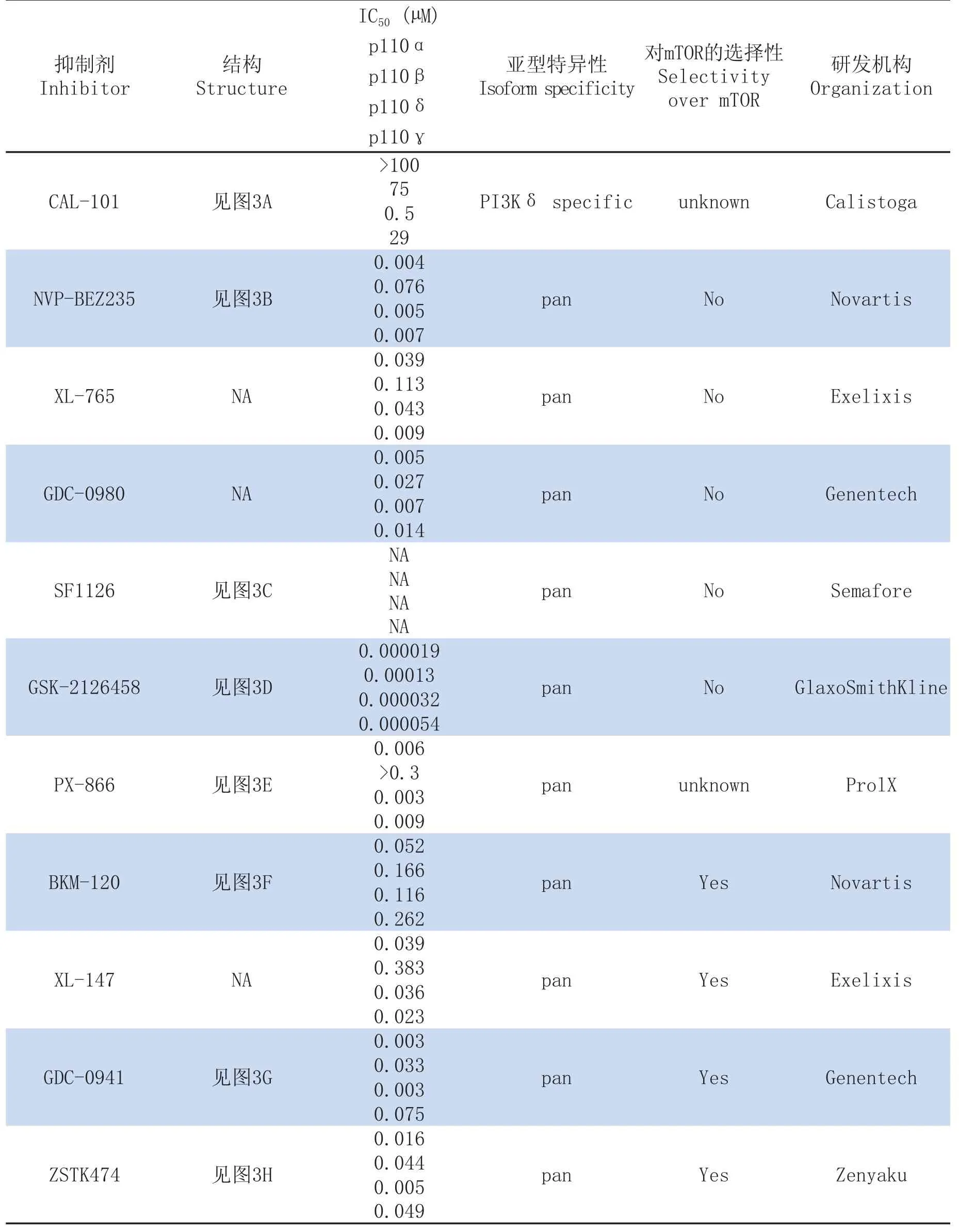
表1 临床试验中的主要PI3K抑制剂[5,7-14]Tab 1 Main PI3K inhibitors in clinical trials[5,7-14]
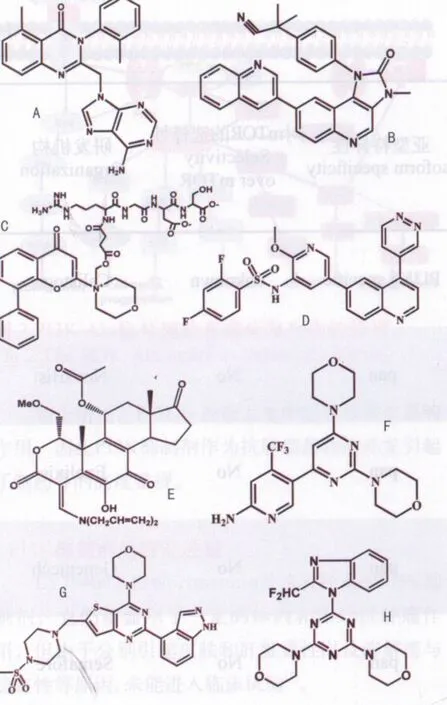
图3 几种PI3K抑制剂的化学结构式Fig 3 The chemical structure of several PI3K inhibitors
3 讨论
关于单纯的PI3K抑制剂与PI3K/mTOR双重抑制剂孰优孰劣的问题一直是从事PI3K研发的制药业界关心的问题。虽然有些研究报告的结论为后者优于前者,理由是在所设计的一些体外试验中后者的抗癌效果更好[15],但这种结论尚需更多和更深入的实验来证明。另外PI3K/mTOR双重抑制剂常常也对DNA-PK有抑制作用[16],这种抑制作用会否产生副作用?虽然动物试验证实了两种类型抑制剂均为安全的,但两者在临床试验中的安全性是否有差异?这些问题还是需要临床试验的最终结果来解答。从我们介绍的进入临床试验的PI3K抑制剂可以发现,几个研发PI3K抑制剂的主要制药公司,包括Novartis、Genentech和Exelixis,不约而同地将两种类型的PI3K抑制剂都推向临床试验,恰恰是因为临床前试验的结果并不能充分证明哪一种类型更好。

A. 对血管内皮细胞血管生成的抑制作用:对血管内皮细胞HUVEC迁移和小管生成的抑制作用;B. 对裸鼠WiDr移植瘤模型的抗肿瘤作用A. Anti-angiogenic effect of ZSTK474 on endothelial cells: the inhibitory activities on HUVEC(human umbilical vein endothelial cells)migration and tube formation. B.In vivo antitumor efficacy of ZSTK474 on WiDr xenograft.图 4 ZSTK474的体内外抗肿瘤作用Fig 4 Antitumor activities of ZSTK474 in vitro and in vivo.
同样一直在争论的问题是泛PI3K抑制剂和PI3 Kα亚型特异性抑制剂孰优孰劣的问题。因为只有编码PI3Kα的基因被发现在癌症中出现变异,出于泛PI3K抑制剂可能出现副作用的考虑,PI3Kα亚型特异性抑制剂的开发一直是一些制药公司的目标。但迄今为止进入临床试验的PI3K抑制剂中尚没有一个真正的PI3Kα特异性抑制剂。考虑到PI3Kβ等已被报道在PTEN缺失的癌症中的作用比α亚型更重要,因此从抗癌效果的角度来考虑,理论上泛PI3K抑制剂比PI3Kα特异性抑制剂应该更好一些,当然这也需要临床试验的结果来证实。
除PI3K抑制剂单独用药外,与其它药物或疗法的联合治疗研究近年内有很多报道。包括与细胞毒类药物的联合用药、与其它分子靶向药物的联合用药以及与放射线疗法的联合治疗等。现在一般认为PI3K抑制剂的主要抗肿瘤作用在于其抑制细胞的生长增殖而不是杀死癌细胞,所以与细胞毒类药物或放疗的联合应用理论上应能增强其抗癌效果。最近的一篇论文报告了ZSTK474与放疗的联合治疗,其效果比单独应用ZSTK474和单独放疗显著提高,且未发现严重的副作用[17]。这种与放疗联合治疗的效果也经NVP-BEZ235的研究得到证明。GDC-0941与细胞毒类药物多西他赛的联合应用也收到较好效果[18]。近年来PI3K抑制剂与分子靶向药物MEK抑制剂的联合应用也有很多报道,包括NVP-BEZ235和GDC-0941等PI3K抑制剂都表现出与MEK抑制剂协同作用的效果[19]。
现在一些PI3K抑制剂如NVP-BEZ235、XL-765和XL-147等的临床I期试验初步结果已经发表,这些化合物的安全性基本上没有问题。随着临床试验的继续深入,其单独以及与其它药物联合用药用于肿瘤治疗的有效性将会被揭晓。让我们一起期待新型分子靶向药物PI3K抑制剂的诞生。
[1] Toker A, Cantley LC. Signalling through the lipid products of phosphoinositide-3-OH kinase[J]. Nature, 1997,387(6634):673-676.
[2] Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers[J]. Science,2004,304(5670):554.
[3] Yuan TL, Cantley LC. PI3K pathway alterations in cancer:variations on a theme[J]. Oncogene, 2008,27(41):5497-5510.
[4] Kong D, Yamori T. Phosphatidylinositol 3-kinase inhibitors:promising drug candidates for cancer therapy[J]. Cancer Sci,2008,99(9):1734-1740.
[5] Kong D, Yamori T. Advances in development of phosphatidylinositol 3-kinase inhibitors[J]. Curr Med Chem, 2009,16(22):2839-2854.
[6] Wendel HG, De Stanchina E, Fridman JS, et al. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy[J]. Nature,2004,428(6980):332-337.
[7] Maira SM, Stauffer F, Brueggen J, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity[J]. Mol Cancer Ther,2008,7(7):1851-1863.
[8] Kong DX, Yamori T. ZSTK474, a novel phosphatidylinositol 3-kinase inhibitor identified using the JFCR39 drug discovery system[J]. Acta Pharmacol Sin, 2010,31(9):1189-1197.
[9] Folkes AJ, Ahmadi K, Alderton WK, et al. The identification of 2-(1H-indazol-4-yl)-6- (4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent,selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer[J]. J Med Chem, 2008,51(18):5522-5532.
[10] Kong D, Yamori T. ZSTK474 is an ATP-competitive inhibitor of class I phosphatidylinositol 3 kinase isoforms[J]. Cancer Sci,2007,98(10):1638-1642.
[11] Kong D, Dan S, Yamazaki K, et al. Inhibition profiles of phosphatidylinositol 3-kinase inhibitors against PI3K superfamily and human cancer cell line panel JFCR39[J]. Eur J Cancer,2010,46(6):1111-1121.
[12] Kong D, Okamura M, Yoshimi H, et al. Antiangiogenic effect of ZSTK474, a novel phosphatidylinositol 3-kinase inhibitor[J]. Eur J Cancer, 2009,45(5):857-865.
[13] Garlich JR, De P, Dey N, et al. A vascular targeted pan phosphoinositide 3-kinase inhibitor prodrug, SF1126, with antitumor and antiangiogenic activity[J]. Cancer Res, 2008,68(1):206-215.
[14] Ihle NT, Williams R, Chow S, et al. Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling[J]. Mol Cancer Ther, 2004,3(7):763-772.
[15] Marone R, Erhart D, Mertz AC, et al. Targeting melanoma with dual phosphoinositide 3-kinase/mammalian target of rapamycin inhibitors[J]. Mol Cancer Res, 2009,7(4):601-613.
[16] Carnero A. Novel inhibitors of the PI3K family[J]. Expert Opin Investig Drugs, 2009,18(9):1265-1277.
[17] Anzai K, Sekine-Suzuki E, Ueno M, et al. Effectiveness of combined treatment using X-rays and a phosphoinositide 3-kinase inhibitor, ZSTK474, on proliferation of HeLa cells in vitro and in vivo[J]. Cancer Sci, 2011,102(6):1176-1180.
[18] Yao E, Zhou W, Lee-Hoeflich ST, et al. Suppression of HER2/HER3-mediated growth of breast cancer cells with combinations of GDC-0941 PI3K inhibitor, trastuzumab, and pertuzumab[J]. Clin Cancer Res, 2009,15(12):4147-4156.
[19] Hoeflich KP, O'Brien C, Boyd Z, et al. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models[J]. Clin Cancer Res, 2009,15(14):4649-4664.
猜你喜欢
中风与神经疾病杂志(2022年9期)2022-10-19
中国生物制品学杂志(2022年3期)2022-05-13
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
海外星云(2021年9期)2021-10-14
皮肤病与性病(2021年3期)2021-07-30
现代仪器与医疗(2021年1期)2021-06-09
大众健康(2020年7期)2020-08-25
爱你(2019年13期)2019-11-14
