
p38 MAPK-HSP27信号通路在内毒素致大鼠肺损伤中的作用*
2012-11-06 08:51:35马涛,刘志
中国病理生理杂志 2012年11期
马 涛, 刘 志
(中国医科大学附属第一医院急诊科,辽宁 沈阳 110001)
1000-4718(2012)11-1943-07
2012-05-16
2012-09-17
沈阳市科技计划(No.F11-264-1-52);中国医科大学附属第一医院科学研究基金资助项目(No.fsfh1103)
△通讯作者Tel:024-83282011; E-mail:liuzhicmu2004@yahoo.com.cn
p38 MAPK-HSP27信号通路在内毒素致大鼠肺损伤中的作用*
马 涛, 刘 志△
(中国医科大学附属第一医院急诊科,辽宁 沈阳 110001)
目的观察p38丝裂原激活蛋白激酶(p38 MAPK)-热休克蛋白27(HSP27)信号通路在急性肺损伤病理过程中的变化规律。方法健康雄性Wistar大鼠(300~320 g)随机分成正常对照组(A组)、急性肺损伤组(B组)及急性肺损伤+SB203580组(C组)。通过腹腔注射内毒素建立急性肺损伤大鼠模型,分别于实验开始后的0、2、4、6、8 h处死各组大鼠。检测支气管肺泡灌洗液(BALF)白细胞介素6(IL-6)、肿瘤坏死因子α(TNF-α)及BALF中蛋白含量。苏木素-伊红(HE)染色检查肺组织病理变化及免疫荧光方法检测内皮细胞内F-actin和G-actin,计算肺湿干重比值(W/D)。检测肺组织中磷酸化p38 MAPK(p-p38 MAPK)及磷酸化HSP27(p-HSP27)的含量。结果B组在实验后2 h BALF中蛋白水平和肺W/D开始明显增加,给予内毒素后8 h肺泡上皮肿胀,肺泡壁增宽,肺泡间质和肺泡腔水肿明显,肺泡内炎症细胞、红细胞和蛋白渗出明显增多,表现出急性肺损伤的病理改变。在给予了p38 MAPK抑制剂SB203580后的C组BALF中蛋白水平及肺W/D分别比B组明显减少,肺泡内炎症细胞、红细胞和蛋白渗出、间质与肺泡水肿均较B组减轻。B组均在实验后2 h血清及BALF中TNF-α和IL-6的浓度开始增加, p-p38 MAPK及p-HSP27的肺内表达开始增加,与A组相比有显著差异。B组实验后8 h的F-actin的表达明显比A组实验后0 h及8 h的增加,给予p38 MAPK抑制剂SB203580的C组肺p-HSP27和F-actin的表达分别比B组明显减少。结论内毒素可以通过激活p38 MAPK-HSP27信号通路引起急性肺损伤;阻断该信号通路可以减轻肺损伤。
急性肺损伤; 细胞因子类; p38丝裂原激活蛋白激酶; 热休克蛋白27; F-肌动蛋白
急性肺损伤(acute lung injury,ALI) 是指机体遭受严重感染、创伤、休克等打击后,出现以弥漫性肺泡毛细血管膜损伤导致的肺水肿和肺不张为特征的病理状态,其最终严重阶段是急性呼吸窘迫综合征(acute respiratory distress syndrome,ARDS)。ALI 的发病机制有很多研究,至今尚未完全阐明。
肺血管内皮细胞衬于血管壁内表面,是血管壁与血液间的分界细胞,其形态结构受损或功能改变导致血管内皮屏障功能的损害,促进急性肺损伤的发生、发展。有研究发现,内皮细胞骨架与血管内皮通透性的调节密切相关,对于维持内皮细胞的屏障功能有重要意义。纤维状肌动蛋白(F-actin)是细胞骨架中的主要收缩蛋白,增加其含量可以使内皮细胞的通透性增加,导致血管屏障功能的损害[1-2]。热休克蛋白27(heat-shock protein 27,HSP27) 作为一种重要的肌动蛋白结合蛋白,在细胞内可被p38丝裂原激活蛋白激酶(p38 mitogen-activated protein kinase,p38 MAPK)信号途径磷酸化,进而参与调节肌动蛋白聚合/解聚过程,使肌动蛋白细胞骨架发生重组,促进急性肺损伤的发生[3-4]。
我们拟通过腹腔注射内毒素(脂多糖,lipopolysaccharide,LPS)建立急性肺损伤大鼠模型,观察p38 MAPK-HSP27信号通路在急性肾损伤诱发急性肺损伤过程中的变化规律,为急性肺损伤的预防、早期诊治提供实验及理论依据。
材 料 和 方 法
1材料
健康雄性Wistar大鼠,体重300~320 g,由中国医科大学动物中心提供。所用仪器包括便携式心电监护仪、高速离心机。5%水合氯醛、大鼠IL-6和TNF-α检测的ELISA试剂盒购自上海森雄科技实业公司。SB203580购自碧云天公司。p38 MAPKⅠ抗、 p-p38 MAPKⅠ抗、HSP27Ⅰ抗和p-HSP27Ⅰ抗均购自Santa Cruz。G-actin染色液(Alexa Fluor® 594 conjugate deoxyribonuclease I)、F-actin染色液(Alexa Fluor® 488 phalloidin)和细胞核染色液(DAPI)均购自Invitrogen。
2方法
2.1动物模型制备和分组 健康雄性Wistar大鼠(300~320 g),予5%水合氯醛300 mg /kg腹腔麻醉,气管切开,颈动、静脉穿刺,留置导管。监测呼吸频率、心率、收缩压、舒张压、平均动脉压及中心静脉压。大鼠的状态稳定0.5 h 后,随机分成3组。A组: 正常对照组,给予腹腔注射生理盐水4 mL;B组:LPS组,给予腹腔注射生理盐水2 mL和LPS(5 mg/kg) 2 mL;C组:LPS+SB203580组,给予腹腔注射p38 MAPK的专一抑制剂 SB203580 2 mL和LPS(5 mg/kg) 2 mL。
2.2标本收集与检测 造模后,分别于实验开始后的0、2、4、6和8 h处死各组大鼠,每个时点处死6只大鼠。所有大鼠处死后留取静脉血标本测定血中细胞因子。开胸暴露两肺,先观察肺脏大体改变,结扎右肺门,取右上肺,用4%多聚甲醛固定;常规石蜡包埋、切片、苏木素-伊红(HE)染色,光镜下检查肺组织病理变化;取右下肺,用滤纸吸去右肺下叶血,称湿重后,置烤箱中(80 ℃、72 h)烤至恒重,称干重,计算肺湿干重比(wet/dry weight ratio,W/D)。左侧肺行支气管肺泡灌洗,每次用3mL的磷酸盐缓冲液(PBS)灌洗左肺,共3次,合并3次收集到的支气管肺泡灌洗液(bronchoalveolar lavage fluid, BALF) 冻存待测细胞因子。用考马斯亮蓝法测定BALF中蛋白含量。
2.3HE染色 肺组织常规固定、脱水、包埋、切片后制作HE切片,每张HE染色切片随机选取多个高倍镜视野,观察并比较肺组织细胞的改变。
2.4IL-6和TNF-α含量的检测 采用双抗体夹心酶联免疫吸附法(ELISA) 检测血清和BALF中的TNF-α和IL-6的浓度。检测步骤按照试剂盒说明书操作。
2.5Western blotting检测p38 MAPK、 p-p38 MAPK、HSP27及 p-HSP27 开胸取出右肺中叶,迅速液氮冻存,制备匀浆,提取蛋白,测定蛋白浓度后,然后保存于-80 ℃待用。取100 μg 的蛋白样品于12% SDS-PAGE 凝胶中进行电泳,待目的蛋白接近凝胶底部时停止电泳,4 ℃、120 V恒压电转移至PVDF膜上,5%脱脂奶粉室温封闭1 h。分别加入1∶1 000稀释的p38 MAPK、 p-p38 MAPK、HSP27及 p-HSP27 Ⅰ抗和1∶500 稀释的GAPDH (内参)Ⅰ抗,4 ℃孵育过夜,加入辣根过氧化物酶标记的Ⅱ抗(1∶1 000)室温孵育1 h后发光显色,凝胶成像系统照相,实验结果采用Quantity One软件分析,扫描吸光度(A)值。
2.6免疫荧光法检测内皮细胞内F-actin和G-actin (1)石蜡标本切片,烘烤2 h左右,脱蜡水化(依次经二甲苯,污水乙醇,95%乙醇,85%乙醇,75%乙醇,PBS);(2)抗原修复(柠檬酸缓冲液高温高压修复);(3)1×PBS缓冲液(0.01 mol/L,pH 7.4)洗涤3次,每次5 min;(4)滴加非免疫山羊血清(内含0.3%Triton X-100)封闭液,覆盖液面2~3 mm,室温放置60 min,甩去多余液体;(5)滴加200 μL F-actin染色(0.165 mol/L)和G-actin染色液(0.3 μmol/L)染色15~20 min;(6)PBS洗3次各5 min;(7)滴加200 μL DAPI染色液染色3 min;(8)PBS洗3次各5 min;(9)荧光显微镜下观察、拍照,F-actin和G-actin分别为绿色和红色荧光,细胞核为蓝色荧光;(10)采用MOTIC FLUO 1.0 分析软件对图片进行区域亮度分析,比F与G肌动蛋白荧光亮度的比值来进行半定量分析。
3统计学处理

结 果
1大鼠BALF中蛋白水平和肺W/D的改变
从表1可以看出,A组在实验后BALF中蛋白水平和肺W/D无明显变化。B组在实验后2 h BALF中蛋白水平和肺W/D开始增加,与A组比较明显差异。C组BALF中蛋白水平和肺W/D比B组明显减少(P<0.05)。
表1大鼠支气管肺泡灌洗液中蛋白水平和肺湿干重比的改变
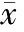
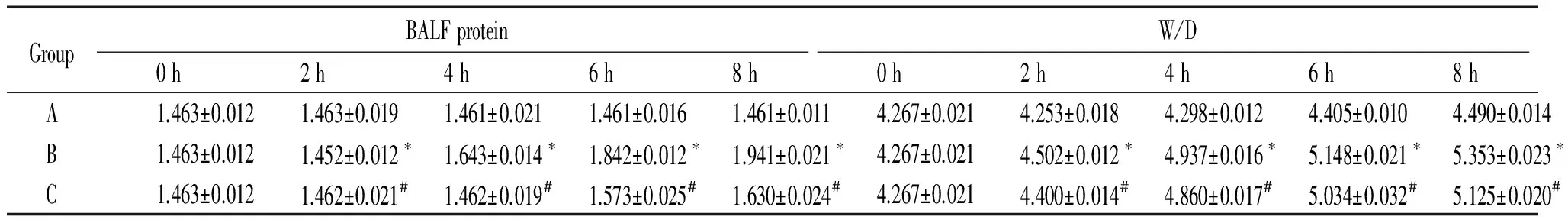
GroupBALFproteinW/D0h2h4h6h8h0h2h4h6h8hA1.463±0.0121.463±0.0191.461±0.0211.461±0.0161.461±0.0114.267±0.0214.253±0.0184.298±0.0124.405±0.0104.490±0.014B1.463±0.0121.452±0.012∗1.643±0.014∗1.842±0.012∗1.941±0.021∗4.267±0.0214.502±0.012∗4.937±0.016∗5.148±0.021∗5.353±0.023∗C1.463±0.0121.462±0.021#1.462±0.019#1.573±0.025#1.630±0.024#4.267±0.0214.400±0.014#4.860±0.017#5.034±0.032#5.125±0.020#
*P<0.05vsA group;#P<0.05vsB group.
2血清和BALF中TNF-α和IL-6的浓度
如表2、3所示,B组大鼠血清和BALF中TNF-α和IL-6的浓度在实验后2 h开始增加,与A组相比差异显著(P<0.05)。C组大鼠血清和BALF中TNF-α和IL-6的浓度在实验后2 h也开始增加,但没有B组增加得更明显,与A、B组相比均有显著差异(P<0.05)。

表2 大鼠血清和支气管肺泡灌洗液中TNF-α的浓度
*P<0.05vsA group;#P<0.05vsB group.
表3 大鼠血清和支气管肺泡灌洗液中IL-6的浓度
*P<0.05vsA group;#P<0.05vsB group.
3LPS和SB203580对大鼠肺病理学变化的影响
正常对照组0 h与8 h肺泡结构完整,肺泡内无明显渗出,无肺间质水肿表现。LPS组肺泡上皮肿胀,肺泡壁增宽、毛细血管扩张和充血,肺泡间质和肺泡腔水肿明显,肺泡内炎症细胞、红细胞和蛋白渗出明显增多,大量炎症细胞浸润,部分视野可见小呼吸道损伤,肺泡结构紊乱,表现出急性肺损伤的病理改变。在给予p38 MAPK抑制剂SB203580后的C组内可见肺泡内炎症细胞、红细胞和蛋白渗出,间质与肺泡水肿均较B组减轻,见图1。
4大鼠p-p38MAPK的表达
B、C组在实验后2 h p-p38 MAPK的条带浓度开始增加,但C组明显比B组浓度减低。半定量分析显示A组在实验后p-p38 MAPK无明显变化。B、C组在实验后2 h p-p38 MAPK开始增加,与A组比较有明显差异(P<0.05)。C组与B组相比明显表达减低 (P<0.05),见图2。
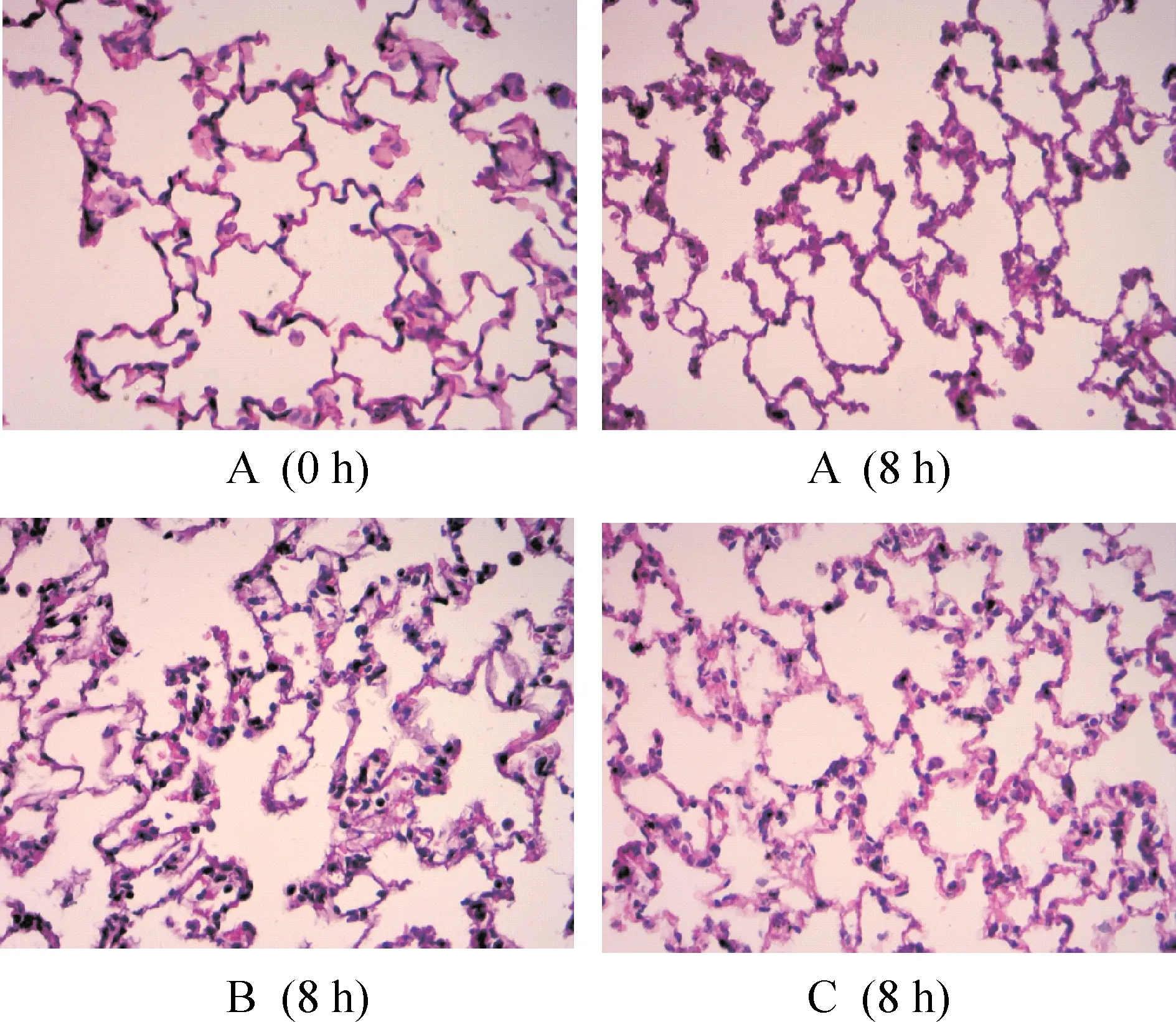
Figure 1. HE staining of rat lung tissues (×400).
图1大鼠肺组织HE染色结果

图2不同时点p-p38MAPK的表达
5大鼠p-HSP27的表达
Western blotting检测发现,B组在实验后2 h p-HSP27的条带浓度开始增加,C组的条带浓度比B组小 。B组在实验后2 h p-HSP27的肺内表达开始增加,与A组相比显著差异(P<0.05)。在给予了p38 MAPK抑制剂SB203580后的C组肺p-HSP27的表达比B组明显减少(P<0.05),见图3。

图3不同时点磷酸化热休克蛋白27的表达
6免疫荧光法检测F-actin和G-actin的表达水平
图4显示B组F-actin的表达比正常对照组0 h及8 h的表达明显增加,p38 MAPK抑制剂SB203580 处理后C组F-actin的表达分别比B组减少。各组免疫荧光检测结果的半定量分析显示,B组均在损伤后2 h F-actin/G-actin比值开始增加,与正常对照组相比显著差异(P<0.05)。给予p38 MAPK抑制剂SB203580后的C组F-actin/G-actin比值比B组明显减少(P<0.05)。
讨 论
内毒素是一些炎症介质和细胞因子的强诱导剂,其致伤作用主要在于它的始动作用。注射内毒素后,其对肺脏的直接损伤作用较少,起致伤作用的主要是它诱发的炎症反应。多种炎症细胞或效应细胞激活后,释放大量炎症介质或细胞因子等,造成肺损伤。本研究发现,通过大鼠腹腔注射内毒素后,大鼠体内炎症介质TNF-α和IL-6的增加,BALF中蛋白含量增加,肺W/D增加,HE染色显示肺泡内炎症细胞浸润、肺泡结构损伤,表现出了典型的急性肺损伤的表现。急性肺损伤是各种直接或间接致伤因素损伤毛细血管内皮细胞及肺泡上皮细胞,造成弥漫性肺间质及肺泡水肿,导致的急性低氧性呼吸功能不全。其基本病理特征是肺部的急性炎症和肺微血管通透性增加[5-7]。肺内皮细胞与上皮细胞屏障完整性与功能受损是急性肺损伤的一个重要特征。细胞的空间结构由细胞骨架维持,细胞骨架是维持细胞正常形态与功能的重要结构。微丝是细胞骨架的一种重要类型,由肌动蛋白构成,主要由F-肌动蛋白和肌动蛋白结合蛋白组成,其聚合/解聚过程与细胞的许多功能活动相关[8]。本研究发现内毒素引起了肺内F-肌动蛋白浓度增加,这说明内毒素引起了肺内皮细胞内F-肌动蛋白开始聚合,应力纤维形成,细胞骨架发现改变。细胞内中心张力增加,细胞收缩,细胞间缝隙形成,肺微血管通透性增加,引起了肺损伤的发生[8-9]。
另外本研究发现通过大鼠腹腔注射内毒素后,引起大鼠BALF中炎症介质TNF-α和IL-6的增加,同时大鼠肺内p-p38 MAPK含量也相应逐渐增加,这表明内毒素引起p38 MAPK的激活可能与TNF-α和IL-6相关[10-11]。同时在实验中也发现如给予p38 MAPK抑制剂SB203580,肺内炎症介质水平也相应减低,这说明p38 MAPK也可能参与调节着炎症介质的激活[3,12]。MAPK是细胞内重要的信号系统,调控细胞增生、分化、凋亡、基因表达等。p38 MAPK是MAPK家族成员之一。研究发现,紫外线照射、促炎细胞因子(TNF-α、IL-6)、低氧、高渗环境、蛋白合成抑制剂、缺血、再灌注以及脂多糖等均可激活p38 MAPK[3,12]。p38 MAPK磷酸化后,调节下游的丝裂原活化蛋白激酶激活蛋白激酶2激酶和HSP27。
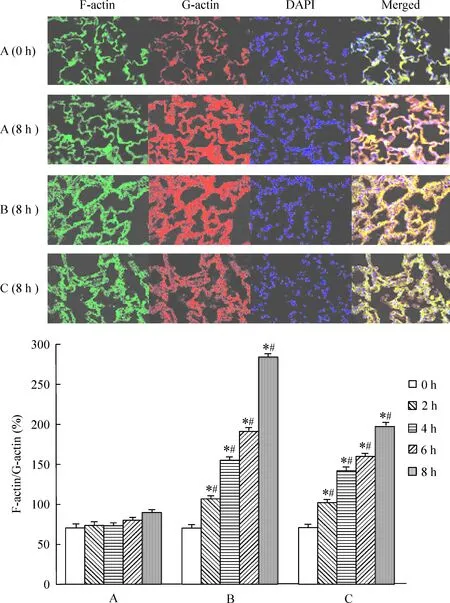
图4肺内皮细胞F-actin和G-actin的表达
本研究发现p- HSP27表达水平随着p-p38 MAPK的增加而增加,呈正相关趋势,且如使用p38 MAPK阻断剂SB203580,则p-p38 MAPK和p-HSP27表达水平下降,这证明p38 MAPK调控着p-HSP27的表达。HSP27作为一种重要的肌动蛋白结合蛋白,在调节肌动蛋白细胞骨架重组方面发挥着重要的作用。现已发现非磷酸化状态存在的HSP27作为帽状蛋白结合在F-肌动蛋白的正极[13],抑制肌动蛋白的聚合。p38 MAPK磷酸化调节下游的MK2 激酶、HSP27,使与F-肌动蛋白正极结合的HSP27在15、78 和82 位的丝氨酸残基发生磷酸化,导致HSP27从F-肌动蛋白正极解离进入胞浆,从而使F-肌动蛋白正极暴露。此时,游离的F-肌动蛋白正极能够与G-肌动蛋白结合, 其结果是F-肌动蛋白发生聚合而延长[14-16],进而使肌动蛋白细胞骨架得以重组,细胞间隙增大,导致肺内皮-上皮细胞屏障功能受损,进而引起急性损伤的发生。本研究发现随着磷酸化HSP27表达水平的增加,肌动蛋白F/G比值、BALF中蛋白浓度及肺W/D也相应增加,肺损伤程度也相应增加。这也证实了磷酸化的HSP27调控肌动蛋白的聚合,促进F-actin延长,延长的F-actin引起了细胞间连接改变,促进肺内皮细胞-上皮细胞屏障完整性与功能受损伤。
我们通过实验发现内毒素引起细胞因子TNF-α和IL-6增加,同时体内p-p38 MAPK、p-HSP27和肌动蛋白F含量也逐渐增加,引起急性肺损伤的发生。如使用p38 MAPK阻断剂SB203580,则p-HSP27表达水平下降和肌动蛋白F含量增加的趋势减缓,同时肺损伤的程度也相应减轻。这可能是由于大鼠腹腔注射内毒素后氧化应激、炎症介质等激活p38 MAPK,之后移位至细胞核内,通过调节下游蛋白HSP27活性而诱导肺血管内皮细胞内F-actin 的再生和重组,使细胞间联系改变,进一步影响通透性。这表明p38 MAPK- HSP27信号通路在大鼠腹腔注射内毒素后致急性肺损伤过程中发挥了重要作用。
[1] 王 彬, 闵 苏.机械牵拉对人肺微血管内皮细胞单层通透性、F-肌动蛋白变化的影响[J].临床心血管病杂志, 2011, 27(1):59-62.
[2] Dudek SM, Garcia JG.Cytoskeletal regulation of pulmonary vascular permeability[J]. J Appl Physiol, 2001, 91(4):1487-1500.
[3] Kiemer AK,Weber NC,Fürst R,et al. Inhibition of p38 MAPK activation via induction of MKP-1:atrial natriuretic peptide reduces TNF-α-induced actin polymerization and endothelial permeability[J].Circ Res,2002, 90 (8):874-881.
[4] Kayyali US,Pennella CM,Trujillo C,et al.Cytoskeletal changes in hypoxic pulmonary endothelial cells are dependent on MAPK-activated protein kinase MK2[J]. J Biol Chem, 2002, 277(45):42596-42602.
[5] Nonas S,Miller I,Kawkitinarong K,et al. Oxidized phospholipids reduce vascular leak and inflammation in rat model of acute lung injury[J].Am J Respir Crit Care Med, 2006, 173(10):1130-1138.
[6] 李 利,方 强,何 非.前列腺素E-1对脂多糖诱导的大鼠急性肺损伤的拮抗作用[J].中国病理生理杂志,2007,23(4):810-812.
[7] 詹丽英,李文澜,柯 伟,等.内毒素急性肺损伤中p38 MAPK、NF-κB与HO-1的关系[J].中国病理生理杂志,2010,26(9):1790-1795.
[8] Dos Remedios CG, Chhabra D, Kekic M,et al. Actin binding proteins: regulation of cytoskeletal microfilaments[J]. Physiol Rev, 2003, 83(2): 433-473.
[9] Zieglerl ME, Souda P,Jin YP, et al. Characterization of the endothelial cell cytoskeleton following HLA class I ligation[J]. PLoS One, 2012, 7(1):e29472.
[10]刘 苏,唐元章,王光磊,等.糖皮质激素通过抑制p38MAPK信号通路缓解大鼠内毒素性急性肺损伤[J].中国病理生理杂志,2009,25(2):338-343.
[11]沈伟锋,赵晓刚,丁 玲,等.盐酸戊乙奎醚抑制脂多糖致急性肺损伤大鼠肺组织p38丝裂原活化蛋白激酶的活化[J].中国病理生理杂志,2009,25(3):551-554.
[12]Jiang Y, Gram H, Zhao M, et al.Characterization of the structure and function of the fourth member of p38 group mitogen-activated protein kinases,p38δ[J]. J Biol Chem, 1997, 272(48): 30122-30128.
[13]Dorion S, Landry J. Activation of the mitogen activated protein kinase pathways by heat shock[J]. Cell Stress Chaperones, 2002, 7(2):200- 206.
[14]Lavoie JN, Hickey E, Weber LA,et al. Modulation of actin microfilament dynamics and fluid phase pinocytosis by phosphorylation of heat shock protein 27[J].J Biol Chem, 1993, 268(32):24210-24214.
[15]Mounier N, Arrigo AP. Actin cytoskeleton and small heat shock proteins: how do they interacte? [J]. Cell Stress Chaperones, 2002, 7(2):167-176.
[16]Gerthoffer WT, Gunst SJ. Invited review: focal adhesion and small heat shock proteins in the regulation of actin remodeling and contractility in smooth muscle[J]. J Appl Physiol, 2001, 91(2):963-972.
Roleofp38MAPK-HSP27signalingpathwayinratacutelunginjuryinducedbylipopolysaccharide
MA Tao, LIU Zhi
(DepartmentofEmergencyMedicine,theFirstHospitalofChinaMedicalUniversity,Shenyang110001,China.E-mail:liuzhicmu2004@yahoo.com.cn)
AIM: To observe the role of p38 mitogen-activated protein kinase (p38 MAPK)-heat-shock protein 27 (HSP27) signaling pathway in lipopolysaccharide-induced acute lung injury (ALI) in rats.METHODSWistar rats were randomly divided into control group, ALI group and ALI+SB203580 group. After the experimental model was established, the rats were sacrificed. The pathological changes of the lung and the changes of F-actin and G-actin in the endothelial cells were observed. The ratio of wet weight to dry weight (W/D) of the lung tissues was measured. The protein levels in bronchoalveolar lavage fluid (BALF) were detected. The levels of IL-6 and TNF-α in serum and BALF were tested. The concentrations of p-p38 and p-HSP27 in the lung were determined.RESULTSIn ALI group, the protein levels in BALF and W/D ratio of the lung increased significantly at 2 h. The levels of TNF-α and IL-6 in serum and BALF began to increase at 2 h, which had significant difference as compared with control group. Aleolar epithelial swelling, alveolar walls widening, alveolar interstitial and cavity edema, and the exudation of alveolar inflammation cells, red blood cells and protein were observed in ALI group. The protein levels in BALF and W/D ratio of the lung in ALI+SB203580 group were much less than those in ALI group. The exudation of alveolar inflammation cells, red blood cells and protein, and the interstitial and alveolar edema in ALI+SB203580 group alleviated as compared with ALI group. The expression of p-p38 MAPK and p-HSP27 in the lung at 2 h in ALI group was higher than that in control group. F-actin expression in ALI group obviously increased than that in control group at time points of 0 h and 8 h. Compared with ALI group, the expression of p-HSP27 and F-actin in ALI+SB203580 group was reduced.CONCLUSIONLipopolysaccharide activates p38 MAPK-HSP27 signaling pathway and induces lung injury. Blockage of p38 MAPK-HSP27 signaling pathway may reduce lung injury.
Acute lung injury; Cytokines; p38 mitogen-activated protein kinase; Heat-shock protein 27; F-actin
R563.8
A
10.3969/j.issn.1000-4718.2012.11.005
猜你喜欢
现代临床医学(2021年4期)2021-07-31 07:55:44
猪业科学(2021年3期)2021-05-21 02:06:18
心肺血管病杂志(2020年5期)2021-01-14 00:43:52
中国畜牧杂志(2020年1期)2020-01-16 04:09:54
国际呼吸杂志(2019年8期)2019-04-29 09:15:12
国际呼吸杂志(2019年8期)2019-04-29 09:15:04
老年医学与保健(2017年6期)2017-02-06 05:29:53
湖南中医药大学学报(2016年1期)2016-12-01 04:08:16
中国医科大学学报(2015年10期)2015-03-01 02:09:59
中国医学科学院学报(2013年6期)2013-03-11 20:26:00
