
固相萃取-高效液相色谱-质谱法检测大豆中氯嘧磺隆除草剂的残留量
2012-10-25 05:26邹月利
食品工业科技 2012年17期
邹月利,陶 波
(东北农业大学理学院,黑龙江哈尔滨 150030)
固相萃取-高效液相色谱-质谱法检测大豆中氯嘧磺隆除草剂的残留量
邹月利,陶 波*
(东北农业大学理学院,黑龙江哈尔滨 150030)
实验建立了固相萃取-高效液相色谱-质谱分析检测方法,对大豆中氯嘧磺隆除草剂的残留量进行了研究。样品经甲醇-超声波提取、正己烷液-液分配、固相萃取柱净化,将处理好的样品进行液相色谱定量分析、采用液相色谱-质谱进行定性检测。实验结果表明:氯嘧磺隆在0.1~200.0mg/L范围内浓度与峰面积具有良好的线性关系,相关系数为0.9997(n=3);在2.0~100.0μg/kg范围内,加标回收率为92.6%~95.2%;RSD为1.27%~2.64%;最低检测限为10.0μg/kg(S/N=3)。本方法具有简便、快速、准确、净化效果好等特点,适用于大豆中氯嘧磺隆除草剂的残留分析检测。
固相萃取,高效液相色谱-质谱法,氯嘧磺隆除草剂,残留检测
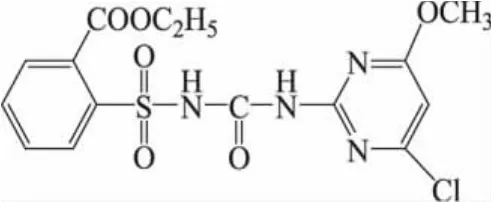
1 材料与方法
1.1 材料与仪器
大豆样品 东北农业大学农学院提供;甲醇、二氯甲烷 进口色谱纯;超纯水 娃哈哈集团有限公司;纯度大于97.4%的氯嘧磺隆原药 大连瑞泽农药有限公司;其它试剂均为分析纯。
Agilent 1100型高效液相色谱仪、Agilent 1100 LC-MSD-TRAP型液相色谱-质谱联用仪 配有自动进样器,紫外检测器,美国Agilent公司;高速万能粉碎机 天津市泰斯特仪器有限公司;R-205旋转蒸发仪 上海申生生物技术有限公司;KL512氮吹浓缩仪 东联仪器设备有限公司;AL-104电子天平
瑞士梅特勒-托利多仪器公司;KQ500DV超声波清洗仪 昆山超声波仪器公司;TGL-16G离心机(最高转速:16000r/min) 上海安亭仪器公司;AP-01P真空抽滤机 天津奥特赛恩斯仪器有限公司;0.45μm微孔滤膜 上海亚东公司;固相萃取柱:Sep-edTMC18;移液枪 德国公司生产。
1.2 实验方法
1.2.1 实验条件
1.2.1.1 色谱条件 采用Agilent-ZORBAX SB-C18色谱柱(4.6×150mm,5.0μm);检测波长为254nm;柱温为室温;进样体积20.0μL;采用甲醇-水(pH2.5)=75∶25作为流动相体系,流速为0.8mL/mim。
1.2.1.2 质谱条件 采用电喷雾电离源(ESI);正(负)离子扫描模式;质量扫描范围 100.0~1000.0amu;干燥气流速 10.0L/min;干燥气温度335.0℃;雾化压力 25.0psi。
1.2.2 大豆样品的处理
1.2.2.1 粉碎 称取约200g的大豆样品,用粉碎机磨碎,混合均匀后备用。
1.2.2.2 提取 准确称取过筛后的大豆粉5.00g,置于50.0mL具塞离心管中,加入甲醇30.0mL,超声波提取20.0min,用离心机离心15.0min(6000r/min),将上清液转移至250mL分液漏斗内。重复3次,合并提取液;向上述提取液中加入30.0mL正己烷-甲醇饱和溶液,振荡2min,静置分层,弃去正己烷,重复3次。将甲醇层用真空抽滤机抽滤2次,去除大豆基质,然后用250mL梨形瓶收集。置于旋转蒸发仪上35℃,80.0r/min减压浓缩至约1.0mL,最后用氮吹浓缩仪吹干,用6.0mL甲醇-水(乙酸调节pH2.5,体积比为 3∶7)溶解,待净化处理[8]。
1.2.2.3 净化 分别用2.0mL甲醇、2.0mL甲醇-水(乙酸调节pH2.5,体积比为3∶7)预淋洗C18固相萃取柱后,在Sep-edTMC18固相萃取柱中加入待净化样品,再向C18固相萃取柱内加入2.0mL甲醇-水(乙酸调节pH 2.5,体积比为1∶1),弃去淋出液。最后分三次共加入15.0mL甲醇-水(乙酸调节pH2.5,体积比为7∶3),收集淋出液,离心 10.0min(12000r/min),将上清液置于旋转蒸发仪上35.0℃,80.0r/min减压浓缩至约1.0mL,然后用氮吹浓缩仪吹干[9]。最后用甲醇溶解、定容至1.5mL,用0.45μm微孔滤膜过滤后用色谱仪进行分析检测。
2 结果与分析
2.1 流动相的选择
实验考察了不同的流动相及其配比:甲醇、甲醇-水、甲醇-水(pH2.5,用甲酸调节)、甲醇-水(pH2.5,用36.0%的乙酸调节)和甲醇-水(pH2.5,用85.0%的磷酸调节)溶液作为流动相体系。结果发现:采用甲醇-水(pH2.5,用甲酸调节)作为流动相时,由于甲酸易挥发会对平行性和分离效果有影响;采用甲醇-水(pH2.5,用85.0%的磷酸调节)溶液作为液相色谱的流动相时基线平稳,色谱峰尖锐,峰形对称,分离效果较好。但是,由于流动相里面有磷酸时会对液质联用仪有影响;采用甲醇-水(pH2.5,用36.0%的乙酸调节)作为流动相体系,流速为0.8mL/mim,甲醇∶水(pH2.5)=75∶25 时也能达到满意的分离效果[10-12]。
2.2 提取溶剂的选择
分别采用二氯甲烷、乙酸乙酯、甲醇-水、甲醇-水(pH2.5,用36.0%乙酸调节)、甲醇-水(pH2.5,用85.0%的磷酸调节)、甲醇和乙腈作为提取溶剂,结果发现:用二氯甲烷和乙酸乙酯提取样品会出现不同程度的乳化现象,高速离心后,提取液仍浑浊不清;另外,用乙酸乙酯提取样品后,大豆中的许多杂质也同时被提取,干扰后续的分析。采用甲醇-水、甲醇-水(pH2.5,用36.0%乙酸调节)和甲醇-水(pH2.5,用85.0%的磷酸调节)的提取方式,均能使磺酰脲类除草剂从大豆基质转移到甲醇中,由于氯嘧磺隆在酸性介质中不稳定,容易分解;同时考虑到水的介入将增加后续净化的步骤,选用甲醇和乙腈作为提取剂,提取率均在92.5%以上。由于乙腈成本较高,同时其毒性较大,所以选择甲醇作为提取溶剂最适合[13-15]。
2.3 标准曲线的绘制
准确称取纯度为97.4%的氯嘧磺隆原药0.1026g,用 250mL烧杯加入少量甲醇溶解,用500.0mL的容量瓶定容至刻度线,得到浓度为200.0mg/L氯嘧磺隆原药储备液。然后配制成浓度依次为 0.1、0.2、0.4、0.6、0.8、1.0、2.0、4.0、6.0、8.0、10.0、40.0、80.0、120.0、160.0、200.0mg/L 的标准溶液,用0.45μm微孔滤膜过滤后,按照1.2.1节色谱条件进行液相色谱测定,得原药的液相色谱图(如图1),保留时间为8.519min。以浓度为横坐标,峰面积为纵坐标,绘制工作曲线(如图2)。结果得到回归方程y=25.851x-2.137,相关系数 r=0.9997(n=3)。表明对照品溶液0.1~200.0mg/L范围内线性良好,可以满足定量分析的要求。
2.4 大豆样品的测定
2.4.1 定量检测 实验对3个品种的大豆进行处理,液相色谱检测,进样量为20.0μL,得到大豆样品的液相色谱图(如图3),数据采用外标法定量(如表1)。实验结果表明,所测大豆粉中氯嘧磺隆的含量为12.7~16.3μg/kg。均低于美国和日本对大豆中氯嘧磺隆除草剂的最低残留限量0.05mg/kg值。
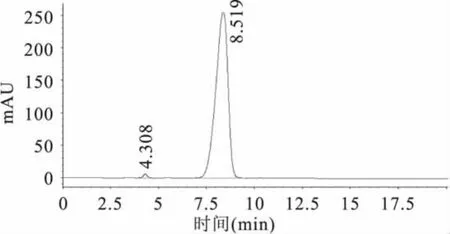
图1 氯嘧磺隆原药的液相色谱图Fig.1 The chromatogram of chlorimuron-ethyl original drug
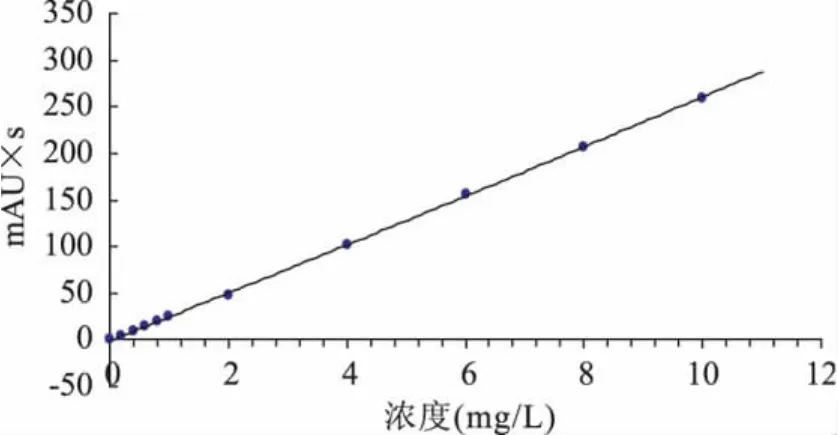
图2 氯嘧磺隆的标准工作曲线Fig.2 The standard working curve of chlorimuron-ethyl
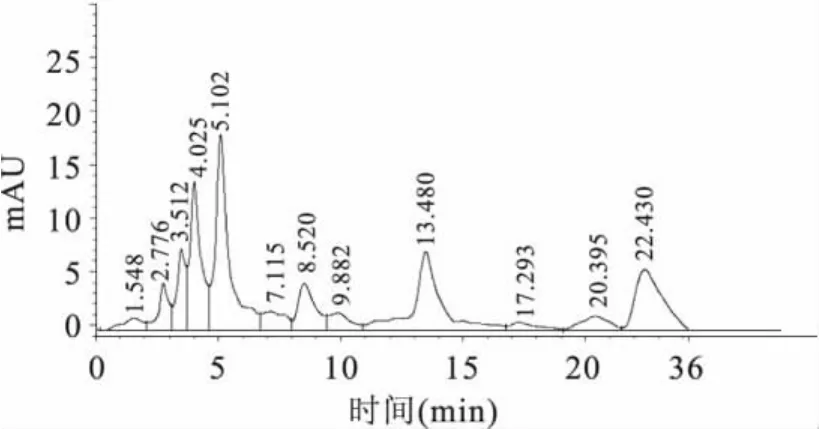
图3 大豆样品的液相色谱图Fig.3 The chromatogram of soybean sample
从图3可以看出,样品中氯嘧磺隆的保留时间为8.520min,和氯嘧磺隆原药的保留时间基本一致,而且样品中氯嘧磺隆与相邻物质的分离效果较好。

表1 3种大豆样品中氯嘧磺隆的残留量Table 1 The residual quantity of chlorimuron-ethyl in 3 kinds of soybean samples
2.4.2 定性分析 实验分别对3种大豆中提取的样品做HPLC-MS分析检测(如图4、图5)。同时根据质谱图解析方法:a.同位素峰,比如含氯的化合物。b.加合离子,比如加钠的就是M+23的峰,或者是自身加合的2M+1,或者2M+23的峰等。c.断裂碎片,离子在源内发生断裂,产生碎片被检测到。d.还有些化合物会脱水。e.基质或者流动相的干扰,在相同条件下被检测到等等[16]。实验结果表明,图4:414.9为M+1,436.8 为 M+23,458.7 为 M+46;图5:413.1 为M-1(M为氯嘧磺隆分子量,Na的分子量为23)。因此,从质谱数据可以进一步对样品中的氯嘧磺隆除草剂进行定性。
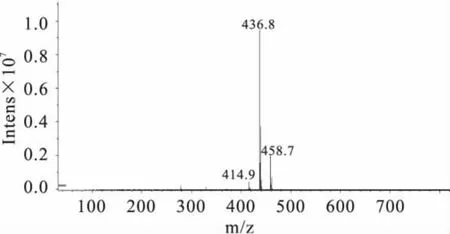
图4 样品中氯嘧磺隆的质谱图(+MS)Fig.4 The mass spectrogram(+MS)of chlorimuron-ethyl in soybean sample
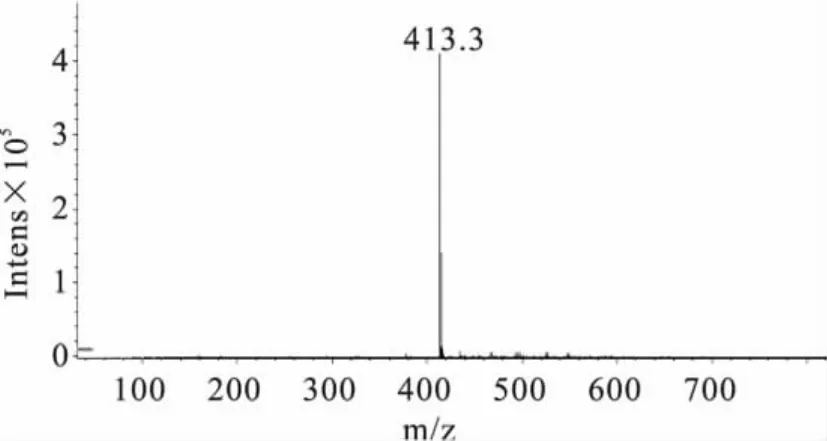
图5 样品中氯嘧磺隆的质谱图(-MS)Fig.5 The mass spectrogram(-MS)of chlorimuron-ethyl in soybean sample
2.5 方法的稳定性和重现性
取同一供试样品溶液,按照1.2节建立的方法进样分析,分别在0、24、48h测定峰面积,相对标准偏差(RSD)为1.43%~2.64%。实验结果表明,供试品溶液在48h内基本稳定。然后分别准确称取同一大豆粉样品3份均5.0g,样品按1.2.2节方法处理,按照1.2.1节色谱条件进样分析,测定氯嘧磺隆含量分别为 12.5、12.6、12.7μg/kg,平均含量为 12.6μg/kg,RSD为0.79%(n=3)。结果表明,方法重现性好。
2.6 方法的精密度和回收率
取同一供试样品溶液,按1.2.1节色谱条件重复进样3次,测定氯嘧磺隆含量分别为13.3、13.5和13.7μg/kg平均值为13.5μg/kg,RSD 为 1.05%(n=3),表明该方法精密度很高。然后向大豆空白样品中添加不同浓度的氯嘧磺隆除草剂标准溶液,按1.2.2节样品处理方法制备供试品溶液,按照1.2.1节色谱条件进样分析,加标回收率92.6%~95.2%,RSD为1.27% ~2.39%。
2.7 方法的最小检测限
实验采用2~3倍噪声为最小响应值,对3种大豆样品种中氯嘧磺隆除草剂残留量进行检测,方法的最低检出限均低于10.0μg/kg(S/N=3),可以满足最低残留限量的检测要求。
3 结论
实验采用了以甲醇作溶剂、超声波提取、正己烷液-液分配、Sep-edTMC18固相萃取柱净化,建立了大豆中氯嘧磺隆残留量的高效液相色谱-质谱测定方法。本测定方法操作简便、快速、准确、净化效果好、灵敏度高等特点,可用于大豆中氯嘧磺隆残留量的定性与定量的分析检测。
[1]祁彦,占春瑞,张新忠,等.高效液相色谱法同时测定大豆中10种磺酰脲类除草剂的残留量[J].色谱,2004,22(6):634-638.
[2]Blasco C,Font G,Manes J,et al.Screening and evaluation of fruit samples for four pesticide residues[J].J AOAC Int,2005,88(3):847-853.
[3]祁彦,李淑娟,占春瑞,等.高效液相色谱-质谱法测定大豆中磺酰脲类除草剂多残留量的研究[J].分析化学,2004,32(11):1421-1425.
[4]Obana H,Okihashi M,Akutsu K,et al.Determination of neonicotinoid pesticide residues in vegetables and fruits with solid phase extraction and liquid chromatography mass spectrometry[J].J Agric Food Chem,2003,51(9):2501-2505.
[5]赵永刚,张祥志,胡冠九,等.固相萃取-高效液相色谱法测定地表水中磺酰脲类农药的研究[J].分析科学学报,2008,24(3):287-290.
[6]叶贵标,张微,崔昕.高效液相色谱-质谱法测定土壤中10种磺酰脲类除草剂多残留[J].分析化学,2006,34(9):1207-1212.
[7]王和兴,黎源倩,雍莉,等.固相萃取-高效液相色谱法同时测定大豆和大米中的磺酰脲类和二苯醚类除草剂残留[J].色谱,2007,25(4):536-540.
[8]Lentza- Rizos C,Avramides E J,Visi E.Determination of residues of endosulfan and five pyrethroid insecticides in virgin of olive oil using gas chromatography with electron-captyre detetion[J].J Chromatogr A,2001,921(2):297-304.
[9]赵进英,辛暨华,郭英娜,等.固相萃取富集-高效液相色谱法分离检测水中3种苯脲除草剂[J].分析化学,2004(7):103-106.
[10]郝征红.固相萃取-HPLC技术在农药残留分析中的研究与应用[D].济南:山东师范大学,2006.
[11]隋凯,李军,卫锋,等.固相萃取-高效液相色谱法同时检测大米中12种磺酰脲类除草剂的残留[J].色谱,2006,24(2):152-156.
[12]祁彦,张新忠,占春瑞,等.高效液相色谱法测定大豆中磺酰脲类除草剂的残留[J].农药,2005,44(2):76-78.
[13]Brown H M.Modes of action,crop selectivity,and soil relations of the sulfonylures herbicides[J].Pestic Sci,1987,29(3):263-281.
[14]Martel A C,Zeggane S.Determination of acaricides in honey by high-performance liquid chromatography with photodiode array detection[J].J Chromatogr A,2002,954:173-180.
[15]任晋,黄翠玲,赵国栋,等.固相萃取-高效液相色谱-质谱联机在线分析水中痕量除草剂[J].分析化学,2001(8):876-880.
[16]Rastrelli L,Totaro K,Simone F D.Determination of organophosphorus pesticide residues in Cilento(Campania,Italy)virgin olive oil by capillary gas chromatography[J].Food Chem,2002,79:303-305.
Determination of chlorimuron-ethyl residue in soybean by solid phase extraction-high performance liquid chromatography-mass spectrometry
ZOU Yue-li,TAO Bo*
(College of Science,Northeast Agricultural University,Harbin 150030,China)
A solid phase extraction-high performance liquid chromatography-mass spectrometry method was established for determination the residue of chlorimuron-ethyl herbicide in soybean.Samples were extracted by methanol with ultrasonic,then liquid-liquid partitioned with normal hexane and purified by solid phase extraction.The sample was quantitatively analyzed by high performance liquid chromatography and qualitatively analyzed by high performance liquid chromatography-mass spectrometry.Results showed that:a good linear range from 0.1~200.0mg/L with correlation coefficient of above 0.9997(n=3)was obtained.The extraction recovery were 92.6%~95.2%and the relative standard deviation were 1.27%~2.64%.The limit of detection was 10.0μg/kg(S/N=3).The described method was simple,sensitive and accurate.
solid phase extraction;high performance liquid chromatography-mass spectrometry;chlorimuron-ethyl herbicide;residue detection
TS207.3
A
1002-0306(2012)17-0291-04
2012-02-27 *通讯联系人
邹月利(1980-),男,硕士,实验师,研究方向:农药降解及残留分析。
哈尔滨市科技攻关计划项目(2007GG002544)。
猜你喜欢
今日农业(2022年16期)2022-11-09
中国化肥信息(2022年5期)2022-08-30
今日农业(2021年20期)2021-11-26
环境保护与循环经济(2021年7期)2021-11-02
今日农业(2021年14期)2021-10-14
食品安全导刊(2021年20期)2021-08-30
今日农业(2019年15期)2019-01-03
现代园艺(2017年19期)2018-01-19
营销界(2015年23期)2015-02-28
河北工业科技(2015年4期)2015-02-27
