
基于稀疏表示算法的蛋白质质谱数据特征选择
2012-10-11 09:21柯激情厉力华郑智国孟旭莉
Biophysics Reports 2012年8期
柯激情, 祝 磊, 厉力华, 韩 斌,郑智国, 孟旭莉
1.杭州电子科技大学生命信息与仪器工程学院,杭州 310018;
2.浙江省肿瘤研究所,杭州 310022
基于稀疏表示算法的蛋白质质谱数据特征选择
柯激情1, 祝 磊1, 厉力华1, 韩 斌1,郑智国2, 孟旭莉2
1.杭州电子科技大学生命信息与仪器工程学院,杭州 310018;
2.浙江省肿瘤研究所,杭州 310022
高维、小样本数据的特征选择方法在蛋白质质谱数据处理分析领域有着广泛应用。本文针对蛋白质质谱特征选择问题,结合稀疏表示这一新理论框架,提出了一种基于稀疏表示的特征选择算法(sparse representation based feature selection,SRFS)。该方法将稀疏表示分类的结果作为评定某一个特征子空间特征相对重要性的度量,然后通过对大量随机采样子空间计算结果的统计,得到特征空间中每个特征的排序,并进一步分析提炼出与肿瘤疾病相关的若干谱峰。通过在卵巢癌公共数据集OC-WCX2a和浙江省肿瘤医院乳腺癌数据集BC-WCX2a上的实验结果表明,SRFS算法可以有效应用于本文所使用的SELDI-TOF蛋白质质谱数据的分析。
蛋白质质谱;稀疏表示;特征选择
引 言
肿瘤治疗的关键在于早期发现、早期治疗,这已成为人们的共识。研究发现,即使在没有任何症状的早期阶段,肿瘤患者的蛋白质水平已经表现出一系列与细胞癌变相关的变化。这些少量的、异常的变化,实质上是细胞癌变的标志。与疾病密切相关的蛋白被称为“生物标志物” (biomarker)。对这些标志物的研究和鉴定,无论对肿瘤的早期发现和诊断,还是对肿瘤的预防和治疗,都具有重要的应用价值。
质谱技术是研究表达蛋白质组学的有力工具,表面加强激光解吸电离-飞行时间质谱(surface-enhanced laser desorption/ionization time-of-flight mass spectrometry,SELDI-TOFMS)是近年来发展起来的一种全新质谱技术。应用该技术已实现了各种疾病相关标志物的临床检测,如乳腺癌[1]、肝癌[2]、肺癌[3]、卵巢癌[4]和前列腺癌[5]等。SELDI-TOF蛋白质质谱是按照粒子质荷比 (mass to charge ratio,m/z)的大小依次排列而形成的图谱,横坐标表示离子m/z,纵坐标为该离子对应的丰度 (intensity)。质谱数据的样本数量一般为几十至上百个 (102),远小于质谱的维度 (104),从模式识别的角度来看,此类数据的分析属于典型的“小样本问题”,这意味着原始蛋白质质谱数据包含有大量的冗余信息。这些冗余信息不仅使得分析算法的计算量增加,而且降低了分类的精度和性能。
近年来,很多研究者采用不同的模式识别和机器学习方法对蛋白质质谱数据进行了分析处理[6]。从模式识别和机器学习的角度看,其主要框架可划分为数据获取、预处理、特征提取和选择、分类决策这四个部分。其中,特征提取和选择为此框架中最为关键的环节,主要包含两类方法:一类称为特征选择 (feature selection),即将不同的蛋白m/z位点看成变量,采用某种筛选机制,保留对最终分类有利的蛋白位点;另外一类称为特征提取 (feature extraction),基本思想是通过某种变换,将原有蛋白位点映射到一些新的变量上去。特征提取方法 (如PCA[7]和小波变换[8]等)所形成的新特征仅能在分类问题上有使用意义,其所得出的特征并非是具有物理意义的生物标志物,因此,针对质谱数据的特征选择是非常有必要的。
稀疏表示 (sparse representation)方法是信号处理领域近年来的一个研究热点,也被称为压缩传感 (compressive sensing)、稀疏采样等。其基本思想是把给定的信号进行适当的变换,使其在变换域上用尽量少的基函数来表示原始信号,目前已经在语音视频编码、生物医学成像、盲源分析、图像融合等高维数据处理方面获得了广泛、良好的应用[9]。
本文针对基于SELDI-TOF蛋白质质谱数据的肿瘤相关特征提取问题,提出一种新的基于稀疏表示的特征选择算法。首先讨论了稀疏表示方法在质谱数据上的分类性能,其次设计了基于稀疏表示分类器的特征选择算法,筛选出与肿瘤判断密切相关的特征位点。在公共数据集OC-WCX2a和浙江省肿瘤医院数据集BC-WCX2a上的实验结果表明,稀疏表示方法可以有效地应用于SELDI-TOF蛋白质质谱数据。
材料与方法
材料
为了验证稀疏表示方法在蛋白质质谱数据分析上的有效性,本文采用如下数据库进行测试。
1)卵巢癌OC-WCX2a公共数据集。该数据由美国食品和药物管理局 (U.S.Food and Drug Administration,FDA)及美国国家肿瘤研究所 (National Cancer Institute,NCI)提供,采用WCX2蛋白芯片阵列和SELDI-TOF技术相结合对人体血清样本进行分析。数据集包含2类蛋白表达信息:癌症患者 (cancer)和正常对照组 (control),其中癌症患者100例,正常对照100例。每例样本包含15 154维质荷比特征。数据库OC-WCX2a采用人工点样操作,已去除基线。数据预处理部分仅需进行重采样和归一化两步。
2)乳腺癌BC-WCX2a数据集。该数据由合作单位浙江省肿瘤医院提供,包含2类样本:癌症患者87例,正常对照39例。每例样本包含86 426维蛋白特征。由于医院采集的数据存在大量的不确定影响因素,数据质量不高,因此对医院数据集采用如下预处理步骤: a)重采样。由医学知识可知蛋白质特征表达区间一般为1 500~10 000(Da),因此选择此区间范围内的数据进行重采样,采样后得10 000个特征点。 b)基线去除。使用局部极小值方法拟合基线[10],然后将基线从原质谱中去除,以去除质谱中伴有的电子和化学噪声。 c)谱峰矫正。为了标定蛋白谱峰,所用的3个基准蛋白位点为4091.1、5908.0和6634.0。d)归一化。将质谱数据进行0-1标准化,即每个质谱样本的最大强度设为1,最小值设为0,从而使得不同的质谱可以在同一范围内进行分析。方法
信号的稀疏表示是指将信号在特定的原子库中进行分解,若原子库中的原子与信号的主要成分相似,则仅需要少数原子的线性组合,就能很好地逼近原始信号。与传统的线性时频表示 (如傅立叶分解、小波分解)不同的是,稀疏表示采用的分解集是特定的原子库,而非正交基,且强调约束条件为使用最少的原子参与表示。如果原子库为训练样本组成的训练集,则这种稀疏表示可以有效地利用训练集中内在的时频结构。
稀疏表示分类方法简介
令第i类别的训练样本Ai=[ai1,ai2,…,aini]∈Rm×n,那么,具有共同类别属性的测试样本yi∈Rm就可以近似地表示成第i类训练样本的线性组合,即

其中xij∈R,aij∈Rm,xi=[xi1,xi2,…,xini]为样本yi在训练样本Ai下的稀疏表示系数,aij为第i类下的第j个样本。
定义一个由l类训练样本集构成的完整样本字典A=[A1,A2,…Al]。则样本yi在字典A下可表示为:

若定义稀疏度为向量中非零元素的个数,则稀疏表示问题可表述为:

但是,l0范数优化问题实际上是一个NP(non-deterministic polynomial)问题,算法复杂度随着问题规模的增长而呈指数增长,在多项式时间内难以求解,甚至无法验证解的可能性。压缩感知与稀疏表示方法的一些最新研究成果[11]表明,如果解x足够稀疏,式(3)则可转化为式(4)中l1范数正则化的极小化问题求出:

式(4)可以用牛顿迭代法进行求解[12]。
对于测试样本y,可以通过优化目标(4)求出它的稀疏表示系数x,即得到测试样本在全局表示下的稀疏表示。然后根据系数在各个类别上关于y的重构误差设计出合理且高效的分类器[13]。
对于每个类别i,令δi是系数向量x∈Rn关于第i类的特征函数。δi仅保留了x上对应于第i类的那些元素,而将其他元素强制为零,从而得到新的向量x^,利用x^可获得测试样本y仅在第i类测试样本参与下的重构y^i:
显然,y与y^i的残差描述了y在第i类下的重构能力,残差越小,则重构效果越好。从模式识别的角度看,样本y将更有可能归属于此类别。因此可利用原始样本对各类的残差大小作为判别准则来对该样本进行归类:
基于稀疏表示方法的特征选择
质谱数据维数巨大,普通的模式分类方法很难直接应用,因此需要进行特征选择(feature selection)操作。特征选择可以有效地减少样本维数和计算量,其目的是从维度为m的特征中选择出数量为q(m>q)的一组最优特征。与特征提取 (feature extraction)不同,特征选择筛选出的特征,其物理意义没有发生变化,即获得的特征为蛋白位点。本文中的特征选择分为两个步骤进行:
a)特征子空间生成
将m维样本在特征维度上无放回地随机划分成s=m/d个组,这个过程称为一次随机分组。在同一个子空间中,每个样本下具有d维特征。在每次随机分组中,每一个特征有且仅有一次出现在某一个子空间A'∈Rd×n中。
b)分类分析
将子空间A'应用于基于稀疏表示的分类器进行分类分析,其分类结果 (如分类率、敏感性、特异性等)实际上反映了特征子空间的相对重要性。依据这些分类结果,可以得到对应于每一个特征的积分值。由于特征子集是随机生成的,为防止结果的随机性带来的偏差,实验需要进行大量的重复随机抽样计算,可获得代表相对重要性的累积值Score,依据Score的排名可得到所需要的具有判别意义的蛋白位点。
特征选择的算法流程图如图1所示,首先将训练集在维度上进行随机分组,得到s个子空间。对于第i(i=1,2,…,s)个子空间,使用t=10倍交叉验证法(10 fold cross validation)在稀疏表示分类方法上进行预测验证:即轮流选择一份数据作为测试集Te,其余为训练集Tr,来进行稀疏表示分类预测,得到此子空间的分类准确率、敏感性、特异性的均值,记为 mAcc(i)、mSen(i)、mSpe(i)。

图1 基于稀疏表示的特征选择算法流程图Fig.1 Flow chartoffeature selection method based on sparse representation
在一次随机分组中,仅对满足式(7)的随机子空间所对应的特征进行积分累加。
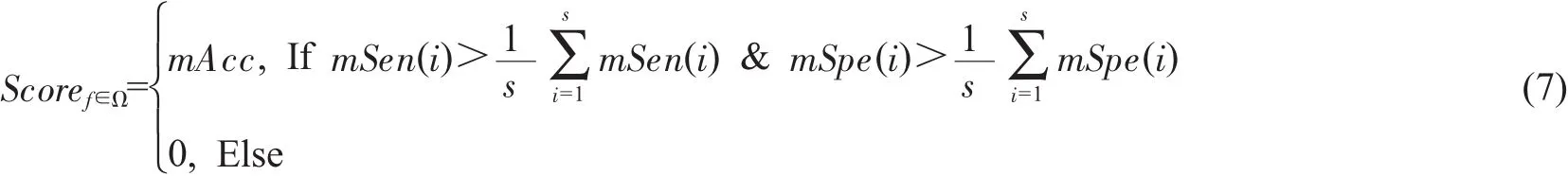
即在每次随机分组中,只有满足特异性、敏感性同时大于平均值时,才对该子空间的特征进行计分累加。累加部分Score的大小取决于该子空间的分类能力,分类能力越高,积分值越大。最终根据多轮随机分组下的总体累加的计分值进行排序,得到最优的特征。
实 验
子空间维度选择实验
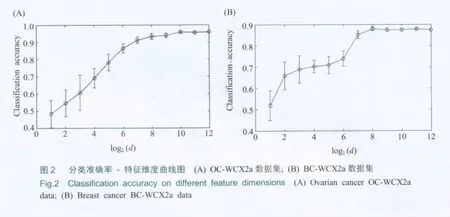
特征子空间的生成中,随机子集维度d的选取至关重要。按照随机子空间生成方法,考察在不同维度d下稀疏表示分类器的分类性能。由10倍交叉验证法 (10 fold cross validation)进行估计,按照9∶1分配训练集与测试集。重复20次实验,得到分类准确率结果均值。稀疏表示分类的分类准确率随随机维度d的变化趋势如图2所示。d的取值为2i(i=1,2,…,12),图2显示了不同维度d下的分类准确率与标准差。由图中可以看出,在OC-WCX2a以及BC-WCX2a数据集上,当d>200时,分类准确率均趋于稳定。因此,在本文的实验条件下,综合考虑计算量和分类准确率,选择特征子集维度d=200。
稀疏表示特征选择
本文实验流程如图3所示,将预处理后的公共数据集OC-WCX2a随机划分成10组,每组20个样本,且每组中两类样本所占比例相同。依次选取其中1组作为验证集,其余9组样本集构成训练集,特征选择部分依据图1所示的SRFS特征筛选流程,重复随机分组1000次,以得到稳定结果。选择前200个最具判别意义的特征,并在验证集上使用SVM进行分类预测,获得不同特征下的分类准确率。在肿瘤医院数据集上,仍按照相同的实验流程,得到不同特征下分类准确率的平均。

图3 实验流程图Fig.3 Flow chart of experiment
结 果
特征选择实验结果
根据常用医学判别标准,实验中设肿瘤样本为阳性,正常样本为阴性,统计真阳性样本数(TP)、真阴性样本数(TN)、假阳性样本数(FP)和假阴性样本数(FN)。采用灵敏性Sen=TP/(TP+FN)、特异性Spe=TN/(TN+FP)及准确率Acc=(TP+FN)/(TP+FP+FT+FN)作为算法性能评价指标。首先将稀疏表示特征选择算法同t-test方法进行比较,分别采用这两种算法挑选出前q=20、50、80、110、140、170和200个特征,采用支持向量机SVM(RBF核)测试所选特征的判别性能。两种方法的训练集与测试集数据划分保持相同。为防止由于随机划分数据集而带来的实验结果的波动,实验中,采取多次试验取均值的方法,分类准确率(Acc)、敏感性(Sen)和特异性(Spe)均采用10次10倍交叉验证 (10 fold cross validation)的平均值作为最终结果。结果如表1所示。

表1 SRCFS与T-test方法的分类性能比较Table 1 Comparison of classification performance on SRFS and T-test
表1中,在OC-WCX2a数据上的实验结果显示,SRFS在选择不同数量的特征时,其判别性能均优于t-test方法。同时也可以看出,两种特征选择方法都取得了较好的分类效果,相对于t-test方法,在各个不同特征点上,SRFS方法的平均分类准确率有0.28%的微弱提升,而另外两个方法得到的分类准确率高达96%。一个可能的原因是由于公共数据集的数据获取过程标准,并且经过了合适的预处理步骤,数据质量较高。
而在浙江省肿瘤医院的乳腺癌数据集BC-WCX2a上的实验结果显示:t-test在20~200个位点的分类率为83.52%~88.87%,与SRFS方法的分类结果 (74.36%~91.20%)相比,均在80个特征点左右达到稳定的分类水平,其中SRFS稳定在92%左右,明显优于t-test的88%。在特征数从20到50变化时,SRFS的特异性出现了大幅度的提升,这可能与医院数据的数据质量及实验的循环参数设置有关,导致对特异性贡献高的特征位点的排名没有进入前20,而位于20到50之间。就医学诊断而言,敏感性即为癌症样本的识别率,特异性即为正常样本的识别率。将癌症患者诊断为正常,会使病人丧失最佳治疗时期甚至导致死亡,因此,在医学诊断中强调更多的是敏感性。
特征位点与蛋白谱峰的对应
由于之前基于SRFS的方法并没有做特征层次的相关性去除,因此,挑选出来的结果含有大量重复和冗余。图4为某次实验筛选出的特征的分布图。可以看到,挑选的特征回溯到原始质谱数据时,所挑选的特征位点分别聚集在某些特定的谱峰附近。从生物标志物的角度来看,这些密集在某一谱峰附近的特征应统一看成是此谱峰的有效表达。在此思路的指导下,将不同训练集下的特征筛选结果经人工精选,得到表达密集的20个点位。
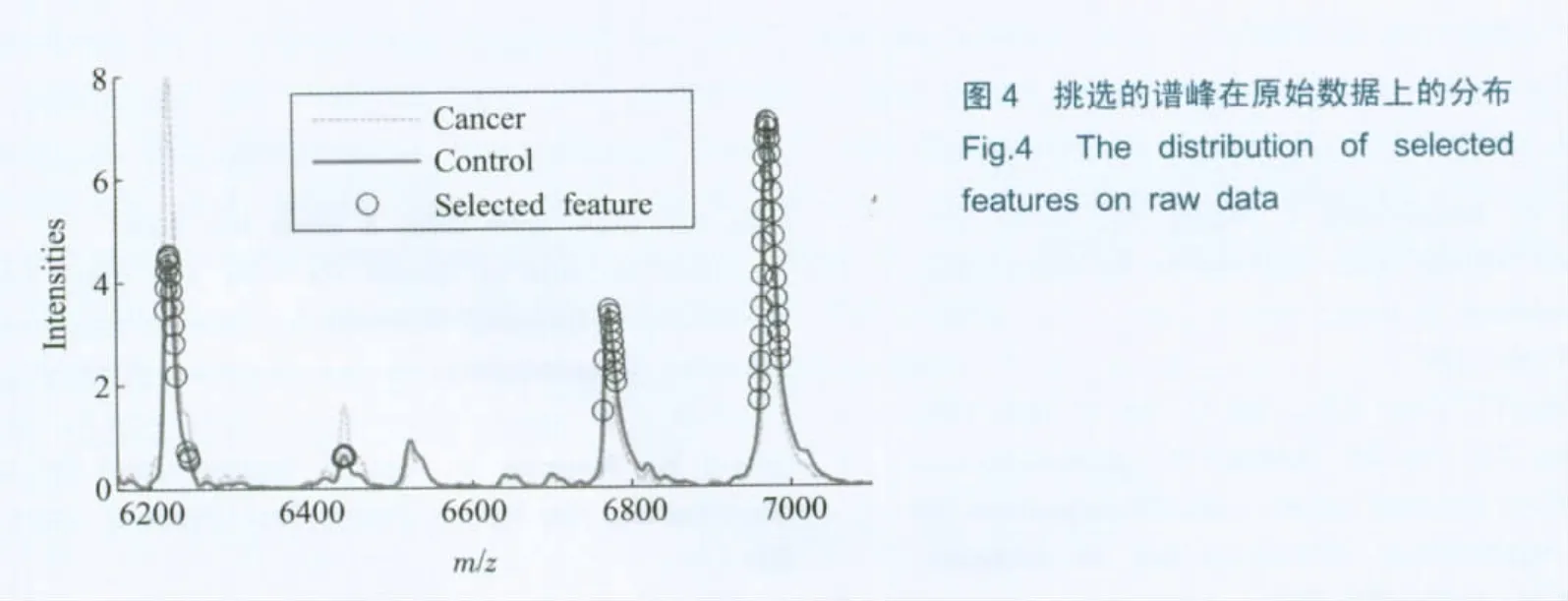
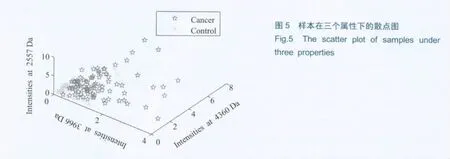
为了进一步得到更精确的点位,在这20个点位中,按照无放回排列组合方式选取3个点,以这些排序作为特征选择的结果,使用SVM在整个数据集上按照10倍交叉验证 (10 fold cross validation)的方式进行分类预测。实验发现,分类准确率结果在0.90以上的实验中,质荷比为2557、3966和6130 Da的这3个点出现的次数最多。其中两类样本在这3个属性下的分布如图5所示。从图中可以看出,在此3个属性上,不同类别样本的分布无明显交叠,各自分布在不同的区域。正常样本聚集在坐标原点附近,而癌症样本分布在外围。在完整的数据集中重新使用交叉验证方法,发现在此3个位点下,在测试集上的分类准确率高达92.12%,敏感性为97.28%,特异性为75.57%。与直接的特征选择结果 (如表1)相比,仅这3个位点就优于特征提取的最好结果91.97%,表明本实验已经挑选出了特征挑选结果中最核心的谱峰。
总 结
本文将稀疏表示方法应用在SELDI-TOF数据的生物标志物筛选中,在公共数据库以及浙江省肿瘤医院临床数据上的实验结果表明,本文算法有较好的性能。特别是在浙江省肿瘤医院的临床数据的实验结果表明,本文算法挑选出的3个质谱谱峰能够准确表达出癌症样本和正常组样本的区别。两类样本在此3个维度的分布已经被明显区分开来。
然而,本文的生物标志物筛选仅依靠了信息学手段,将分类准确率、敏感性和特异性等作为标准,所挑选的标志物在临床上是否有效,有待下一步的临床实验验证。
1. Li J,Zhang Z,Rosenzweig J,Wang YY,Chan DW.Proteomics and bioinformatics approaches for identification of serum biomarkers to detect breast cancer.Clin Chem,2002,48(8):1296~1304
2.Poon TCW,Yip TT,Chan ATC,Yip C,Yip V,Mok TSK,Lee CC,Leung TW,Ho SK,Johnson PJ.Comprehensive proteomic profiling identifies serum proteomic signatures for detection ofhepatocellularcarcinoma and itssubtypes.Clinl Chem,2003,49(5):752~560
3.Zhukov TA,Johanson RA,Cantor AB,Clark RA,Tockman MS.Discovery of distinct protein profiles specific for lung tumorsandpre-malignantlunglesionsby SELDImass spectrometry*1.Lung Cancer,2003,40(3):267~279
4. Tang KL,Li TH,Xiong WW,Chen K.Ovarian cancer classification based on dimensionality reduction for SELDI-TOF data.BMC Bioinformatics,2010,11(1):1~8
5.Larkin SET,Zeidan B,Taylor MG,Bickers B,Al-Ruwaili J,Aukim-Hastie C,Townsend PA.Proteomics in prostate cancer biomarker discovery.Expert Rev Proteomics,2010,7(1):93~102
6. Saeys Y,Inza I,Larraaga P.A review of feature selection techniques in bioinformatics.Bioinformatics,2007,23(19):2507~2517
7.Kirby M.Geometric data analysis:An empirical approach to dimensionality reduction and the study of patterns.New York,NY,USA:John Wiley&Sons,Inc.2000
8. Vannucci M,Sha N,Brown PJ.NIR and mass spectra classification:Bayesian methods for wavelet-based feature selection.Chemometrics Int Lab Systems,2005,77(1-2):139~148
9.Huang K,Aviyente S.Sparse representation for signal classification.Adv Neural Inform Proc Systems,2007,19:609
10.Coombes KR,Tsavachidis S,Morris JS,Baggerly KA,Kuerer HM.Improved peak detection and quantification of mass spectrometry data acquired from surface-enhanced laser desorption and ionization by denoising spectra with the undecimated discrete wavelet transform.Proteomics,2005,5(16):4107~4117
11.Donoho DL.For most large underdetermined systems of linear equations the minimal l1-norm solution is also the sparsest solution.Commun Pur Appl Math,2006,59(6):797~829
12.Kim SJ,Koh K,Lustig M,Boyd S,Gorinevsky D.An interior-pointmethod for large-scale l1-regularized least squares.IEEE J Select Top Signal Proc,2007,1(4):606~617
13.Wright J,Yang AY,Ganesh A,Sastry SS,Ma Y.Robust face recognition via sparse representation.IEEE Transac Pattern Analys Mach Intellig,2008,31(2):210~227
Sparse Representation Based Feature Selection for Mass Spectrometry Data
KE Jiqing1,ZHU Lei1,LI Lihua1,HAN Bin1,ZHEN Zhiguo2,MENG Xuli2
1.College of Life Information Science&Instrument Engineering,HangZhou DianZi University,HangZhou 310018,China;
2.Zhejiang Cancer Institute,Zhejiang Cancer Hospital,Hangzhou 310022,China
This work was supported by grants from the National Natural Science Foundation of China(60801054,60801055),the National Science Foundation for Distinguished Young Scholars of China(60788101),the Zhejiang Province Welfare Technology Applied Research Project(2010C33017),Zhejiang Medical and Health Science Research Fund(2010KYA041),Zhejiang Provincial Science and Technology Projects(Y2080586)
Aug 30,2011 Accepted:Feb 10,2012
LI Lihua,Tel:+86(571)87713525,E-mail:lilh@hdu.edu.cn
Feature selection method has been widely used for protein spectrometry data which has high dimension and small samples size.In this paper,a novel feature selection method based on sparse representation(SRFS)is proposed.SRFS considers a feature be important or informative if the subset containing it can perform well in a sparse representation classifier(SRC).In this method,the relative importance of a subset was measured via SRC.And by means of the results of abundant random subsets,we ranked all the features.We also extracted a few peaks which were related with cancer closely.To investigate the performance,the proposed method was tested and evaluated on the ovarian cancer database OC-WCX2a and breast cancer database BC-WCX2a which supplied by Zhejiang Cancer Hospital.The experimental results show that SRFS can be used to select highly predictive representative feature sets in SELDI-TOF protein spectrometry data analysed in this paper.
Protein mass spectrum;Sparse representation;Feature selection
2011-08-30;接受日期:2012-02-10
国家自然科学基金项目(60801054,60801055),国家杰出青年科学基金项目(60788101),浙江省公益性技术应用研究项目(2010C33017),浙江省医药卫生科学研究基金项目(2010KYA041),浙江省省级科技项目(Y2080586)
厉力华,电话:(0571)87713525,E-mail:lilh@hdu.edu.cn
Q61
10.3724/SP.J.1260.2012.10089
DOI:10.3724/SP.J.1260.2012.10089
猜你喜欢
上海金属(2021年6期)2021-12-02
食品安全导刊(2021年20期)2021-08-30
昆明医科大学学报(2021年3期)2021-07-22
烟草科技(2021年6期)2021-06-24
电脑知识与技术(2018年19期)2018-11-01
自动化学报(2017年5期)2017-05-14
真空与低温(2017年1期)2017-03-15
电子制作(2017年23期)2017-02-02
智能系统学报(2015年4期)2015-12-27
振动工程学报(2014年4期)2014-03-01

- Biophysics Reports的其它文章
- 钙网蛋白的免疫生物学活性研究进展