
代谢网络的重构
2012-10-10 12:10邓世果吴干华杨会杰
上海理工大学学报 2012年6期
邓世果, 吴干华,2, 杨会杰
(1上海理工大学 管理学院,上海 200090;2华南师范大学 南海学院,佛山 528225)
系统生物学的一个重要任务是认识细胞内调控和代谢关系.代谢网络依据基因组数据将代谢系统表示为一个图,为代谢组学数据的分析提供可视化的研究平台.随着基因组测序技术的迅速发展,到目前为止,包括细菌、古生菌、真核生物在内的数百种生物全基因组序列先后测序完成.根据基因组注释信息可以按功能的不同将基因分成不同的组,其中较大的组是可指导合成酶的基因组.酶可催化细胞内代谢反应,依据代谢反应可构造代谢网络.人们构建了专门的代谢反应数据库,如KEGG、BioCyc、PUMA2等,这些数据可以用于构建代谢网络,并且可以对代谢网络作进一步分析.作为复杂非线性动态调控系统,代谢网络的研究越来越受到重视[1].
基因组范围的代谢网络包含的代谢物很多,传统的方法(如代谢控制)不太实用.Ma等[2-3]首次提出具有可操作性的图论方法用以构造和分析代谢网络,得到普遍认可,成为当前对代谢系统的整体结构进行分析的主要手段.构造的代谢网络有3类:用节点表示代谢物,用节点间的连线表示代谢反应,构成代谢物网络;以代谢反应为节点,以节点之间连线表示代谢物,就是反应网络[4-5];以反应所需的酶为节点,以节点间连线表示代谢物,得到酶网络[6-8].以上构建代谢网络方法的基本步骤是:a.从KEGG数据库取得某一个特定物种细胞内全基因组注释信息,得到所有可能的酶;b.根据酶催化的代谢反应数据库,得到选取的物种细胞内所有可能的化学反应,确定出这些化学反应的底物和产物;c.确定出每个化学反应的反应方向;d.将每个化学反应的反应物和底物用有向边连接起来,得到代谢物网络.其它类代谢网络可以采取相似的办法得到.
上述步骤中的关键问题和需要深入考虑的问题包括:a.网络节点的确定.因为很多生化反应涉及到水、氧气、三磷酸腺苷(ATP)、二磷酸腺苷(ADP)等物质,如果在代谢网络中保留这些物质的话,大部分的节点之间通过这类节点就彼此相连了,整个网络将是以这些节点为核心的致密结构,核心致密的网络结构掩盖了人们感兴趣的网络结构.同时,ATP和ADP等只是在反应中提供能量,并不参与物质的合成和分解.因此,这类物质应该去掉,以凸显代谢网络的非平凡结构.有些文献依据经验,简单地将节点度(也就是与其它节点联结的数目)大于10的节点删除掉;有些文献则将节点按照度排序,将排在前20位的节点删除掉;有些文献比较保守,保留了这些度大的节点,同时也列出了这些特殊节点作为分析代谢网络的参考[9].b.生化反应方向的确定.Ma等[2]提出的方案中,系统地列举了多种情况,如消耗氧气、产生二氧化碳等,认为这些化学反应是不可逆的.并且对整个化学反应库进行了系统的鉴别.但是,这一方法本身需要经验的累计,不可避免地有人为因素起作用.特别是他们的工作是在本世纪初完成的,而近10年来代谢数据库不断地进行更新,需要有更具可操作性的代谢网络的构造方法.本文在对前人文献总结的基础上,以构建鱼腥藻代谢网络为例,试图对这两个关键问题提出一些可行的解决方案.
1 代谢数据库
通过基因组注释信息可以识别出能指导合成催化代谢反应的酶的基因.到目前为止出现了多种用于预测物种特异的酶基因、酶、以及酶催化反应的方法,由此产生了许多优秀的代谢数据库,如表1所示.这些数据库是代谢网络重建的必要资源[10-12].

表1 常用生物数据库Tab.1 Common biological databases
2 图论术语
在图论中,图表示元素与元素之间的二元关系,其中,元素表示为图的顶点,元素之间的关系表示为顶点之间的连线.1个无向图G= (V,E),由顶点集合V和边集合E构成,每条边代表1个顶点对(u,v)间的无方向连线.1个有向图D= (V,A),由顶点集合V和弧集合A构成,其中,每条弧代表1个顶点对(u,v)间从u到v的有向边.如果忽略其中所有弧的方向,则一个有向图就成为无向图[13].复杂系统诸多元素作为节点,元素之间的关系作为边,构造出来的图就是复杂系统的复杂网络描述,如描写代谢系统结构的代谢网络.
3 重构代谢网络
3.1 代谢图数据
3.1.1 反应数据
KEGG数据库中文件reaction.list包含迄今发现的所有生化反应.每个反应都有各自的编号,以R开头后跟5位数字;每个化合物也都有各自的编号,以C开头后跟5位数字.如化学反应R00480:
L-Aspartate+ATP=4-Phospho-L-aspartate+ADP
在reaction.list文件中表示为R00480:
C00049+C00002→C03082+C00008
生物体内的很多代谢反应都是有方向的,不可逆性是代谢反应的一个本质特点.而KEGG并没有给出这些信息,Ma和Zeng整理并提出了11种不可逆反应[2]:
a.有氧生成的反应;
b.大多数有二氧化碳生成的反应;
c.大多数有氨气生成的反应;
d.大多数有磷酸盐生成的反应;
e.由S-Adenosyl-L-methionine生成 S-Adenosyl-L-homocystine,提供一个甲基的反应;
f.有四氢叶酸生成,转移一个碳原的反应;
g.大多数消耗ATP,且没有其它高能代谢物(GTP、CAP等)参与的反应;
h.消耗UDP-sugar,转移糖的反应;
i.消耗 CDP-diacylglycerol,转移磷脂酰基的反应;
j.类似于PAPS+A=PAP+B的反应;
k.几种水解反应.
然后Ma和Zeng又对所有的生化反应进行了判别,给出化学反应的方向.
实际上,文献[2]中对代谢途径有着更加全面深入的研究,对代谢途径中发生的化学反应方向等有着明确的结论,这些信息保存在KEGG数据库的reaction_mapformula.lst文件中.这一文件收集了文献中可以找到的生化反应的方向、化学反应发生所在的代谢途径等信息.因此,本文采取一个新的解决方案,即可以采取一个组合的策略确定生化反应的方向:a.在reaction_mapformula.lst里边能查到的化学反应,其反应方向采用这里边已经明确的反应方向;b.剩余的化学反应,能采用Ma等提出的11条规则判断反应方向;c.前两个策略不能判断的其它化学反应,设为双向的[2,14-15].
3.1.2 反应与酶的关系
从KEGG数据库提取酶与反应的有关信息,见“reaction”文件,其中,给出了每个反应以及催化该反应所需的酶的信息,酶都有各自的EC编号,以EC开头,后跟4个整数,整数之间用点隔开,4个整数依次对酶的功能类别进行诠释,越后面的整数对酶的功能描述得越详尽.如R00480:2.7.2.4,表示EC2.7.2.4催化反应R00480.
3.1.3 酶与基因的对应关系
KEGG数据库中含有enzyme文件,enzyme文件对酶的相关信息描述得非常详尽,包括名称、参与生化反应过程和方式、所在的代谢路径、不同物种中对应的基因名称、数据来源等.可由该数据得到某一特定物种里边存在的所有的酶.
3.2 初步代谢图
连接初步代谢反应网络图的具体做法为:对每个反应,以代谢物和代谢底物为节点,以反应为有向边,从代谢物指向代谢底物.如化学反应R00480:
C00049+C00002→C03082+C00008
如果不可逆,从该反应可获得4条有向边:
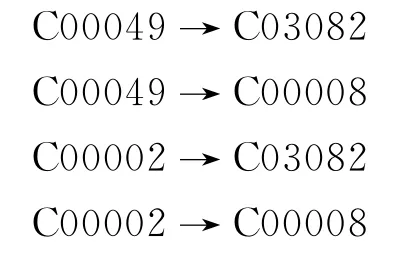
如果可逆,则可将反应拆成方向相反的两个反应,方法同上.把所有反应都转化一遍后就构建完成了一个初步代谢图,以糖酵解过程为例,如图1所示(见下页).
3.3 代谢图的修正
在图1(b)中可以发现蓝色框中的代谢物(CO2、H2O2等)的度比较大,称为通用代谢物(currency metabolite)[4].然而这些通用代谢物一般是电子转移或某些功能基团(磷酸基、氨基、一碳单位、甲基等)转移的携带者,只是协助代谢底物生成代谢产物,并未参与代谢产物的合成.包含有通用代谢物的代谢网络表现出与真实生物意义不符的结论,使得代谢网络的平均最短路径缩短[5,8].如从草酸到氧气只需要两步反应(见下页图1),显然这在细胞内是无法完成的,与细胞内的真实生化反应不符.
因此,为了使细胞中主要化合物的转化表现得更明显,在代谢网络中确切地显示生化反应的步骤,人们通常将这些通用代谢物及一些小分子化合物(如水、氨气、二氧化碳、氧等)从代谢网络中移出.现介绍一种对此问题的处理方法.
文献[2]在KEGG代谢反应数据库的基础上进行手工修正补充后得到一个新的数据库,去掉了每个反应中的通用代谢物及小分子化合物,并明确地给出了每个反应的可逆性信息.在这个数据库中,他们并不是统一地将通用代谢物都去掉,而是根据每个反应来确定其中的通用代谢物.例如,在许多反应中,谷胺酸(glutamate,GLU)和α-酮戊二酸(2-oxoglutarate,AKG)都是用于转移氨基的通用代谢物,然而在反应中AKG参与了合成GLU,因此
AKG+NH8+NADPH→GLU+NADP++H2O+AKG在此反应中它们都是主要代谢物,它们之间的连接应保留[9].由于该数据库几乎全部用手工构建,因此,质量有所保证,已被许多研究者采用.但是,他们的处理依据经验,带有人为性质,并且也已经有近10年的历史,面临数据更新问题.
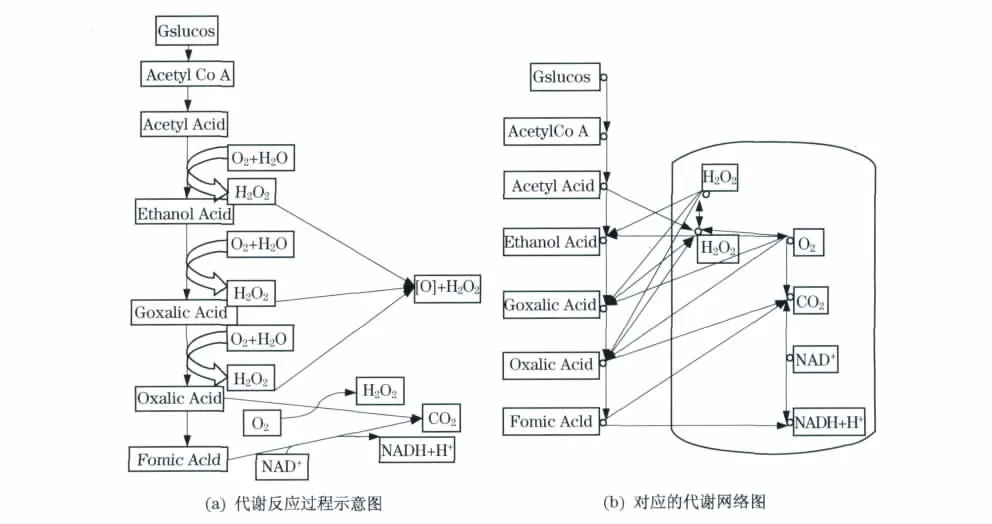
图1 糖酵解过程Fig.1 Glycolysis process
因此,本文提出另外一个可行的办法.在KEGG数据库的LIGAND部分包含的文件reaction_mapformula.lst中给出了代谢路径中的化学反应,即包含了每个反应的方向以及主要代谢物,而省略了如ATP、NADH这一类的辅助因子(cofactor).确定这些主要代谢物采用的标准和文献[2]使用的标准是相同的.上述采用的标准只是针对代谢路径中存在的化学反应进行了处理,而不是所有的化学反应都处理过.因此,本文中采用的策略:a.在reaction_mapformula.lst中出现的化学反应,采用该文件中的信息;b.对比reaction_mapformula.lst与reaction中相同的化学反应,确定出所有可能的通用代谢物.将这些通用代谢物从a不能判断的化学反应中剔除掉.通过以上方法修正后,之前的R00480:

就从4条边减少为只有1条边:C00049→C03082这是因为C0002和C0008均为通用代谢物,这2个节点和与之相连的边都要去掉.这样就可以得到一个完整的代谢物网络,可在此基础上开展进一步的研究,如网络结构、动力学、代谢功能之间的关系的讨论等.
3.4 反应图
图2为将代谢物网络图转化为反应图的示意图.反应图就是以反应为节点,如果两个反应(反应A和反应B)共用一个或多个代谢物或代谢底物,且反应A得到的产物是反应B的底物,就将这两个反应连起来,由A指向B,这样就构成了一个有向的反应图,如图2(c)所示.相对代谢图而言,反应图离酶图关系更近了些.具体细节和需要注意的问题与代谢图类似.
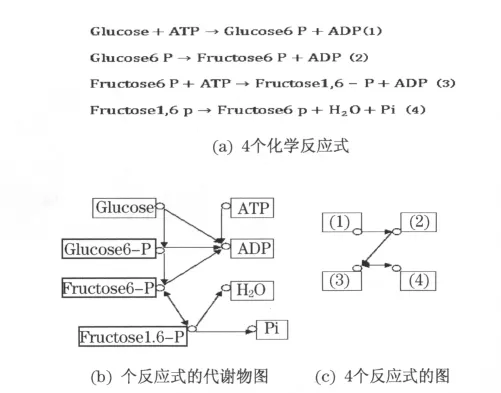
图2 代谢反应网络图Fig.2 Metabolic reaction network
3.5 酶 图
酶图与反应图也并非一一对应,因为,一种酶可以催化不同的反应,一种反应也可能需要多种酶共同参与.酶图以酶为节点,以酶A和酶B为例,如果酶A参与的反应产生的产物正好是酶B参与的反应的底物,则就连接酶A与酶B,且由酶A指向酶B.
4 以鱼腥藻代谢网络为例的网络图
通过以上代谢网络的构造方法,构造出鱼腥藻的代谢网络,并对该网络进行了社团划分,如图3所示,当分为23个社团时,聚类系数Q值最大,此时Q=0.799.
为了更清楚地看清网络结构,图3没有显示度是1的节点,也删除掉了2个孤立的节点.不同颜色表示不同的社团结构.
本文计算了网络度分布(图4)和平均聚类系数.由图4可知,该网络的度分布De服从幂律分布,幂律指数α=-3.0,其聚集系数C(k)与节点度k的关系如图5所示,因此,不满足,C(k)~k∝k-α,即该网络不存在层次结构.经计算,该代谢网络的网络直径为D=21,平均最短路经约为8.从图6(见下页)可知,最可几分布Me发生在最短路径d=7的位置,最可几概率为0.12.以上结论与一般生物代谢网络的性质大体上一致,因此,该重建代谢网络的构造方法是可行的.

图3 鱼腥藻代谢图Fig.3 Topological structure for anabaena metabolic network

图4 鱼腥藻代谢网络的度分布Fig.4 Degree distribution of anabaena metabolic network
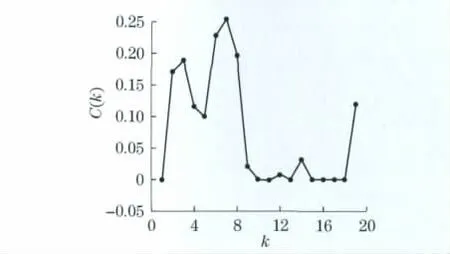
图5 平均集聚系数与度的关系图Fig.5 Relationship of average clustering coefficient versus degree

图6 最短路经分布Fig.6 Distribution function for shortest paths
5 结 论
代谢网络重建是系统生物学的基本任务,是采用复杂网络观点分析代谢数据的基础.随着数据的日益增多和不断更新,设计合理可行的代谢网络重建方法成为迫切需要解决的课题.
本文在总结前人工作基础上,就构建代谢网络步骤中的两个核心问题,提出了可行的解决方法.依据KEGG中文件reaction_mapformula.lst提供的化学反应的方向信息,提出确定代谢网络中边的方向的规则;并且与reaction文件中的化学反应比较,提出确定通用代谢物的规则,用于确定代谢物网络里的节点.以鱼腥藻为例,对构造出来的代谢网络进行了讨论.
代谢网络的重建是一个复杂的过程,也因研究目的的不同而不同.本文讨论的方法适合于复杂网络拓扑结构及其与代谢功能相关性分析.当讨论代谢网络动力学,特别是代谢网络上的流问题的时候,则需要更加准确的网络结构,必须考虑到化学反应在细胞内发生的位置信息、特定环境和细胞生命周期中基因表达量等.因此,高精度的代谢网络涉及脱氧核糖核酸DNA测序和注释、化学反应位置确定、基因表达、化学反应方向、通用代谢物确定等多个复杂环节,是一个复杂的系统工程.本文的构造方法也可用于构造人际关系等网络.
[1]Kanehisa M.Post-genome informatics[M]:Oxford:Oxford University Press,2000.
[2]Ma H W,Zeng A P.Reconstruction of metabolic networks from genome data and analysis of their global structure for various organisms[J].Bioinformatics,2003,19(2):270-277.
[3]Ma H W,Zeng A P.The connectivity structure,giant strong component and centrality of metabolic networks[J].Bioinformatics,2003,19(11):1423-1430.
[4]Wagner A,Fell D A.The small world inside large metabolic networks[J].Proc R Soc Lond B,2001,268(1478):1803-1810.
[5]Light S,Kraulis P,Elofsson A.Preferential attachment in the evolution of metabolic networks[J].BMC Bioinformatics,2005,6(1471):159-169.
[6]Horne A B,Hodgman T C,Spence H D,et al.Constructing an enzyme-centric view of metabolism[J].Bioinformatics,2004,20(13):2050-2055.
[7]Heymans M,Singh A K.Deriving phylogenetic trees from the similarity analysis of metabolic pathways[J].Bioinformatics,2003,19(suppl_1):138-146.
[8]Light S,Kraulis P.Network analysis of metabolic enzyme evolution in Escherichia coli[J].BMC Bioinformatics,2004,5(1):15-20.
[9]Huss M, Holme P.Currency and commodity metabolites:their identification and relation to the modularity of metabolic networks[J].IET Systems Biology,2007,1(5):280-285.
[10]Goto S,Nishioka T,Kanehisa M.LIGAND:chemical database for enzyme reactions[J].Bioinformatics,1998,14(7):591-599.
[11]Maltsev N,Glass E,Sulakhe D,et al.PUMA2——grid-based high-throughput analysis of genomes and metabolic pathways[J].Nucl Acids Res,2006,34(suppl_1):369-372.
[12]Karp P D,Ouzounis C A,Moore-Kochlacs C,et al.Expansion of he BioCyc collection of pathway/genome databases to 160genomes[J].Nucl Acids Res,2005,33(19):6083-6089.
[13]Bondy J A,Murty U S R.Graph theory with applications[M].London:Macmillan,1976.
[14]Guimera R,Amaral L A N.Functional cartography of complex metabolic networks[J].Nature,2005,433(7028):895-900.
[15]Zhao J,Tao L,Yu H,et al.Bow-tie topological features of metabolic networks and the functional significance[J].Chinese Science Bulletin,2007,52(8):1036-1045.
猜你喜欢
现代临床医学(2022年4期)2022-09-29
云南化工(2021年6期)2021-12-21
科学(2020年2期)2020-08-24
少儿美术(2019年1期)2019-12-14
农药科学与管理(2019年5期)2019-08-13
小哥白尼(趣味科学)(2018年6期)2018-09-14
中学生数理化·高一版(2018年6期)2018-07-09
中学生数理化·高一版(2017年10期)2017-12-19
生物技术通报(2015年1期)2015-04-10
质谱学报(2015年5期)2015-03-01
