
纳米锑颗粒的制备及其分散性研究
2012-09-12 07:06徐建林杨树华高威顾玉芬张建斌
航空材料学报 2012年1期
徐建林, 杨树华, 高威, 康 昭, 顾玉芬, 张建斌
(1.兰州理工大学甘肃省有色金属新材料重点实验室,兰州 730050;2.兰州理工大学有色金属合金及加工教育部重点实验室,兰州 730050;3.北京航空材料研究院,北京 100095)
金属纳米材料的制备、结构功能特性及其实践应用的研究成为科研人员共同关注的前沿课题。锑元素与磷元素一样,位于元素周期表的第Ⅴ主族,也是一具有极压、抗磨特性的元素。稀有金属作为添加剂被广泛应用于钢铁工业、催化领域、半导体领域、航空领域、电子产业以及高技术产品等领域,是高附加值产品生产的必须元素,尽管用量不大,却起着其它元素不可替代的作用。作为稀有金属的一员——锑,其具有一系列独特的物理化学性质,纳米锑作为添加剂得到了广泛的应用,在表面涂层材料[1],电极材料[2],纳米晶材料[3],电阻材料[4],化学敏感材料[5],光电材料[6],热敏材料[7]等方面具有广阔的应用前景,已经成为国内外高附加值新型材料研究的热点之一,但有关纯纳米锑颗粒的制备目前还没有专门报道,只在相关文献中见到一些有关纳米锑的制备方法,且都是通过化学反应来合成的[8,9],因此开展纳米锑颗粒的制备技术研究对纳米锑材料的应用以及锑的市场开拓具有积极作用。本文以电化学方法电沉积制得纳米锑(Sb)颗粒,该法操作简单,成本低廉,易于规模生产,颗粒的粒径和形貌易于控制。同时,对所制的纳米锑颗粒在润滑油中的分散稳定做了研究和探讨。
1 实验方法
1.1 电化学制备方法
以纯锑为阳极,不锈钢网为阴极,两极间的距离为30~35mm。将浓HCl,一次蒸馏水和OP-10按一定的比例混合,用磁力搅拌器进行搅拌30min,再将其超声5min,得到试验所用的电解液。采用直流电源进行电解实验。控制电解时间、电流密度以及OP-10的添加量等,制备出了所需的纳米锑颗粒。将电沉积产物经洗涤、干燥,获得了黑色粉末状的纳米锑粉。实验所用直流电源为JW-Z型直流稳压稳流电源,搅拌装置为DF-101S型集热式恒温磁力搅拌器,超声仪为KQ-250E型超声波清洗器,干燥设备为DZF-6050干燥箱。
1.2 纳米锑颗粒的表征
将制备所得的纳米锑适量加入无水乙醇,并进行超声分散,使其均匀分散于无水乙醇中,取一滴滴于铜网上,进行TEM观察,确定微粒的粒径大小与分散性,观察粒子的形貌。将所得纳米锑进行XRD图谱的测定,分析其物相。取适量纳米锑与KBr混合压片,然后进行FTIR谱图的测定,分析其表面改性特性。实验所用设备为D8ADVANCE型X射线衍射仪,JEM-2010型透射电镜,Nexus670FT-IR型傅里叶变换红外光谱仪。
1.3 分散性测定
将制备出的纳米锑颗粒分别以0.1%,0.5%和1%(质量百分比)的添加量加入到纯液体石蜡油中,并用超声分散仪对其超声分散30min,使锑颗粒均匀地分散到纯液体石蜡中,让其在室温下自然沉降。在沉降不同时间之后用分光光度计测其吸光度,实验所用设备为UV-2100型分光光度计。
2 结果与分析
2.1 形貌观察与分析
在制备纳米锑粉时,试验温度为室温(控制在25℃),电极之间的距离为30mm,所用电解液成分均为同一厂家生产,搅拌速度设为恒定值,超声时间为5min,实验环境等条件均保持恒定,改变乳化剂OP-10的添加量、电流密度大小、反应时间,制备出了不同形貌和不同大小的单分散纳米锑颗粒,如图1所示。
图1a所示为OP-10添加量为2ml、电流密度为0.08 A/cm2、反应时间为30min所制备的纳米锑颗粒。图1b所示为OP-10添加量为2ml,电流密度为0.08 A/cm2、反应时间为60min所制备的纳米锑颗粒。由图1a,b可以发现,在其他试验条件相同下,反应时间由30min变为60min而导致纳米锑颗粒的平均粒径由10nm变为80nm,即所制的纳米锑颗粒,随反应时间的延长,其颗粒粒径变大。随着反应时间的增加,生成的Sb3+浓度升高,由于布朗运动和渗透压力的作用,Sb3+穿过界面进入OP-10所形成的微反应器中产生晶核并且不断长大[10],随着纳米锑粒子逐渐长大,OP-10所形成的微团也不断长大,达到一定程度时,OP-10的量不足以形成足够大和足够多的微反应器来容纳所生成的Sb3+,致使所制备的纳米锑颗粒随反应时间的增加而变大,且在某种程度上存在团聚现象,即电化学制备纳米锑颗粒时具有时间效应。
当反应时间为60min,OP-10的添加量为3ml,电流密度为0.08 A/cm2时,制备出的纳米锑颗粒如图1c所示。此时,纳米锑颗粒为不规则多边形,平均粒径30nm,与图1b所示的纳米锑颗粒相比,其他实验条件均没有改变,仅将其OP-10的添加量由2ml变为3ml。在增加了OP-10的添加量后,由于有充足的OP-10形成所需的微反应器来容纳生成的Sb3+,所以所生成的纳米锑颗粒没有进一步的长大,即粒径较小。同时纳米锑颗粒的形貌受电解溶液与表面活性剂的摩尔比值R的影响而形成不规则的多边形[10]。此结果与图1b和图1c所示结果一致。
图1d所示为电流密度为0.04A/cm2,OP-10的添加量为3ml、反应时间为60min所制备的纳米锑颗粒。其形貌是棒状锑颗粒与球状锑颗粒粘到一起所形成的混合体,棒状锑颗粒的平均直径为30nm,长为300nm,球状锑颗粒的平均直径为100nm。其形貌的变化也主要是受电解溶液与表面活性剂的摩尔比值R的影响所致[10]。锑颗粒粒径的变化原因主要是,一方面,由于OP-10的添加量所致,即所生成的纳米锑颗粒的粒径随OP-10的添加量增加而变小,另一方面,由电流密度所决定,即当电流密度小于某一特定值时(小于0.05A/cm2时),会使锑颗粒异常的长大[11]。图1c,d所制备的纳米锑颗粒相比较,其他试验条件均一致,只是将其电流密度由0.08 A/cm2降低到 0.04A/cm2,由图可以看出,电流密度是导致纳米锑颗粒粒径变化的又一因素。本研究中的大量实验表明,电流密度在 0.01~0.004A/cm2范围之间时,锑颗粒的沉积速度与电流密度成线性关系,可获得微米级的锑颗粒;当电流密度在0.05~0.5A/cm2范围之间时,锑颗粒的沉积速度与电流密度偏离直线关系,此时可获得纳米级的锑颗粒,即电化学制备纳米锑颗粒时具有电流效应。这种电流效应与电化学制备其他纳米材料时出现的电流效应一致[11,12]。
2.2 物相分析
图2a~d所示分别为图1a~d四种锑颗粒的XRD衍射图谱。图中各衍射峰的d值依次为3.11,2.25,2.15,这些峰与金属锑的特征峰相吻合。
图2a~c的衍射图谱中只存在锑的特征峰,而无其他物相的特征峰,这表明所制备的产物均为纯净的锑颗粒,无其它杂质。这是由于采用电化学法制备锑颗粒时,发生了如下反应:
阳极:Sb-3e-→Sb3+
阴极:2H++2e-→H2,Sb3++3e-→Sb
溶液中Sb3++3Cl-→SbCl3
由上述反应过程可以看出,Sb3+在阴极被还原成锑原子时,同时有氢气生成,氢气在反应过程中,可以保护电解液中新生成的锑不被氧化,这为得到纯净的纳米锑颗粒提供了有利条件。

图1 纳米锑颗粒的TEM照片Fig.1 TEM photographs of nanometer Sb particles
图2b,d的衍射图谱中,除了具有明显的锑的(012),(104)和(110)晶面衍射峰,同时还出现了较弱的(110),(002),(112),(022)和(132)晶面的衍射峰。出现SbCl3的衍射峰的主要原因是,在这两种工艺条件下制备锑颗粒时,由于OP-10的添加量不足和较低的电流密度,致使锑颗粒粒径较大[10,11],从而导致 OP-10 未能完全地包覆所形成的纳米锑颗粒,使其混有反应过程中所生成的SbCl3的成分,这就使其XRD衍射图谱中出现了SbCl3晶面的衍射峰,由于混有的SbCl3量较少,所以其衍射峰较弱。这一现象与TEM所表征的结果一致,SbCl3的出现也再一次证明了在电解液中确实发生了Sb3++3Cl-→SbCl3这一反应。
由图2a~d还可明显地看出各衍射峰出现了宽化现象,其主要原因是由于纳米锑颗粒细化的结果。根据XRD的衍射数据并以(012),(104)和(110)晶面衍射峰为基准,通过Scherrer公式[13]可计算出纳米锑颗粒的平均粒径,图2a~c中计算结果分别为10nm,80nm和30nm,这与图1a~c的TEM照片所显示的球形纳米锑颗粒和不规则多边形纳米锑颗粒的平均粒径相一致。而根据图2d的XRD衍射数据计算出的平均粒径为30~100nm,结果存在比较大的变动范围,这也证实了该锑颗粒粒径分布不均匀,而是由大小不同的锑颗粒混合构成,如图1d所示。
2.3 红外光谱分析
图3所示为OP-10的红外光谱,从图3中可以看出,氢键缔合的O-H伸缩振动在3550-3200cm-1处有一宽而强的吸收峰,在560cm-1处为C—OH面外弯曲振动吸收峰,在2920cm-1附近的吸收峰是长链烷基的吸收,在1640cm-1附近的峰尖而弱,这表明了OP-10中存在苯环,而且在1100cm-1处的C—O—C震动吸收峰也说明了OP-10的结构。
图4a~d所示为OP-10改性后的纳米锑颗粒的红外光谱,即分别为图1a~d四种锑颗粒的红外光谱。与图3相比,其各吸收峰都在不同程度上减弱,且四个红外光谱图的各特征吸收峰基本一致,这说明OP-10对不同的纳米锑颗粒具有相同的改性方式。经过OP-10改性后的纳米锑颗粒在3440 cm-1附近有—OH伸缩振动吸收峰的存在,且强度减弱,说明OP-10在改性纳米锑颗粒时,羟基起了部分作用,但并不是完全依靠羟基与纳米锑颗粒表面进行化学吸附,同时在2880cm-1附近出现了甲基的对称伸缩振动,这表明在对纳米锑颗粒表面进行改性的过程中,OP-10的长链烷基分子结构没有失去,同时证明在纳米锑颗粒中有OP-10乳化剂的存在。在1640cm-1附近的苯环吸收峰,再次证明了OP-10在纳米锑颗粒中的存在。—CH3以及—CH2—特征吸收峰在吸附前后的变化,说明OP-10通过烷基疏水链与疏水性的锑纳米颗粒表面发生作用,OP-10吸附在纳米锑颗粒的表面,C—H伸缩振动受到抑制,特征吸收峰强度减弱。同时,由于OP-10的其他各基团的特征吸收峰强度也在相当程度上减弱,这表明各基团之间受到了相互抑制作用。综上所述,纳米锑颗粒的表面改性是通过OP-10的长链分子结构与纳米锑颗粒之间的化学吸附以及OP-10的长链烷基分子之间的氢键、范德华力相互作用,分子链相互缠结,部分通过C—H键互相渗入,最终有效地包覆在纳米锑颗粒表面来达到其表面改性效果。各试样在1100cm-1处的C—O—C震动吸收峰的消失,表明在表面改性过程中醚键起了一定的作用。图4a和图4b中,在2360cm-1处出现了新的峰,这可能是由于OP-10和锑原子的共轭结构所引起,而图4c和图4d中该峰不太明显,可能是其吸收峰强度太弱所致。
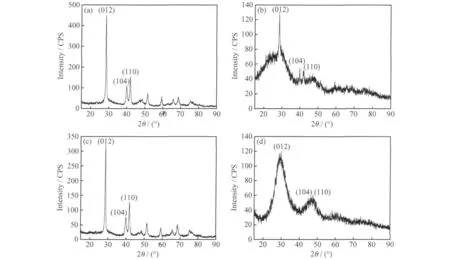
图2 纳米锑颗粒的XRD照片Fig.2 XRD pattern of nanometer Sb particles
2.4 稳定性分析
图5所示为分别含有0.1%,0.5%和1.0%(质量分数)的纳米锑颗粒纯液体石蜡的吸光度(A)随静放时间(T)的变化趋势。根据吸光度的变化,以A-T曲线来表征悬浮液的分散稳定性,悬浮液分散稳定性越好,聚沉越少,A越大。由图可知,含有纳米锑颗粒的纯液体石蜡的吸光度随静放时间的变化规律基本一致,在前24h内,液体石蜡中的纳米锑颗粒迅速沉降,在24h之后,其沉降速度明显减慢。由图5可见,在整个实验过程中,纳米锑颗粒添加量为0.5%的液体石蜡的吸光度(A)均高于添加量为0.1%和1.0%的液体石蜡的吸光度(A)。固体颗粒在液相中的分散稳定主要受两方面因素影响,即固体颗粒与液相介质的润湿作用和在液相中颗粒间的相互作用[14]。本研究中所用纳米锑颗粒和液体石蜡均一致,因此纳米锑颗粒与液体石蜡介质的润湿作用可以忽略不计,其中起主要分散稳定作用的为液体石蜡中纳米锑颗粒间的相互作用。在液体石蜡中,纳米锑颗粒之间的主要作用为布朗运动所产生的碰撞作用以及其受到的重力作用,当添加量为1.0%时,由于浓度较高,在布朗运动的作用下,颗粒间碰撞的几率较大,导致其大量的团聚,从而沉降比较明显,当添加量为0.1%时,由于其浓度较低,颗粒之间的作用较弱,导致其最终沉降的原因主要是重力场作用,而添加量为0.5%时,由于布朗运动碰撞,在没有出现团聚的同时又克服了部分重力的作用,使得其与重力场作用达到了一个平衡状态,从而使其表现出了最佳分散稳定性能。
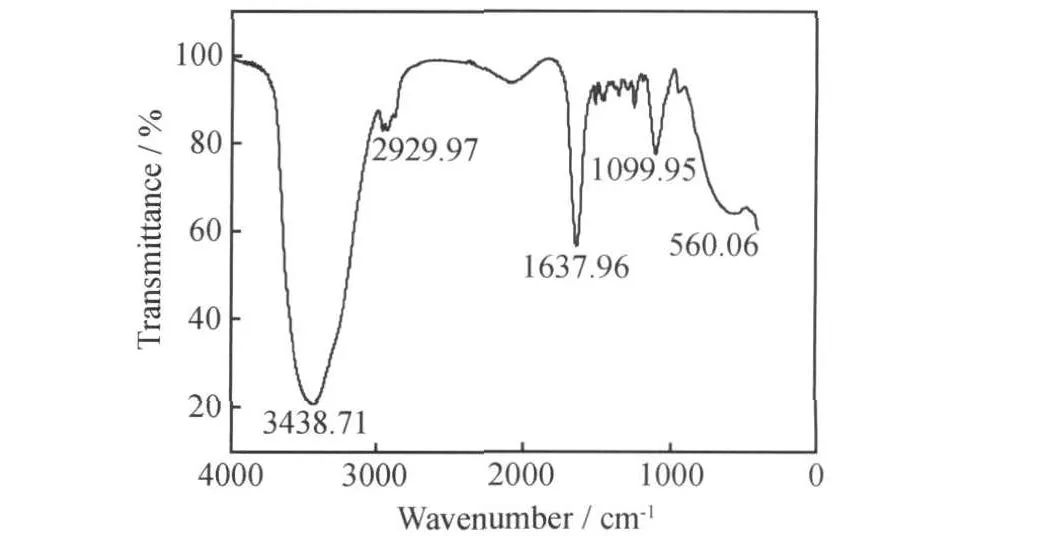
图3 OP-10的红外光谱图Fig.3 The IR spectra of OP-10
3 结论
(1)以电化学方法电沉积制得了不同形貌和粒径的纳米锑颗粒,并且在制备过程中进行了原位表面改性,其中锑颗粒在长大过程中,随电解时间、电流密度和OP-10添加量的不同而得到了不同的纳米锑颗粒。
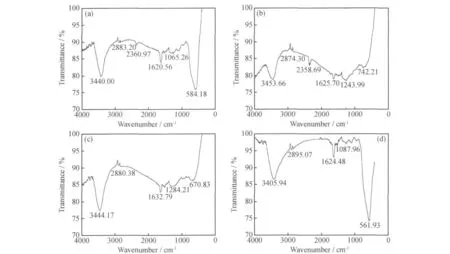
图4 OP-10修饰后的纳米锑颗粒的红外光谱Fig.4 The IR spectra of Sb nanoparticles modified by OP-10
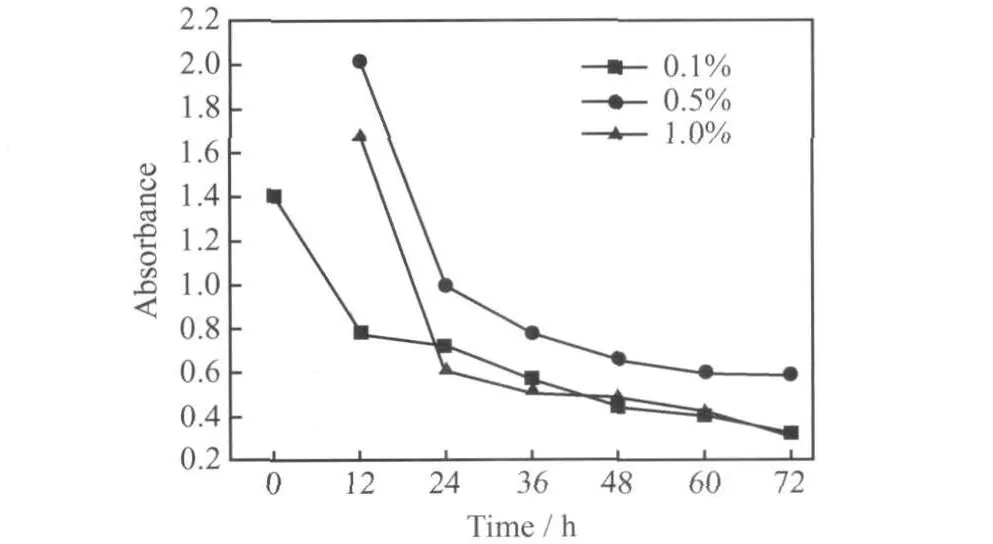
图5 含纳米颗粒的液体石蜡吸光度随时间的变化Fig.5 The changes of absorbance with time
(2)纳米锑颗粒的制备具有时间效应和电流效应。随着反应时间的延长,纳米锑颗粒粒径变大,且在某种程度上存在团聚现象,在一定范围内,适当增加电流密度有利于纳米锑颗粒的形成。
(3)纳米锑颗粒的表面改性,主要是通过OP-10的长链分子结构与纳米锑颗粒之间的化学吸附以及OP-10的长链烷基分子之间的氢键、范德华力相互作用,分子链相互缠结,部分通过C-H键互相渗入,最终有效地包覆在纳米锑颗粒表面来达到其表面改性效果,同时在反应过程中醚键也起了一定作用。
(4)添加量为0.5%的纳米锑颗粒在纯液体石蜡油中的分散稳定性能最佳。
[1]SOM Tirtha,KARMAKAR Basudeb.Surface plasmon resonance and enhanced fluorescence application of singlestep synthesized elliptical nano gold-embedded antimony glass dichroic nanocomposites[J].Plasmonics.2010,5(2):149-159.
[2]TARIQ Fawad,AZHER S.Umair,NAZ Nausheen.Failure analysis of cast lead - antimony battery grids[J].Journal of Failure Analysis and Prevention.2010,10(2):152-160.
[3]RAJGARHIA Rahul K,SPEAROT Douglas E,SAXENA Ashok.Molecular dynamics simulations of dislocation activity in single-crystal and nanocrystalline copper doped with antimony[J].Metallurgical and Materials Transactions(A).2010,41(4):854 -860.
[4]RAVICHANDRAN K,PHILOMINATHAN P.Analysis of critical doping level of sprayed antimony doped tin oxide films[J].Journal of Materials Science:Materials in Electronics.2011,22(2):158 -161.
[5]ZVYAGIN AA,SHAPOSHNIK AV,RYABTSEV SV,et al.Determination of acetone and ethanol vapors using semiconductor sensors[J].Journal of Analytical Chemistry,2010,65(1):94-98.
[6]JAIN Garima,KUMAR R.Electrical and optical properties of tin oxide and antimony doped tin oxide films[J].Optical Materials.2004,26:27-31.
[7]COLEMAN J P,LYNCH A T,MADHUKAR P,et al.Antimony-doped tin oxide powders:Electrochromic materials for printed displays[J].Solar Energy Materials and Solar Cells.1999,56(3-4):375-394.
[8]YADAY Neelam,NAGAR Meena,BOHAR Rakesh.New sol-gel precursors for binary oxides of antimony,Sb2O3(senarmonite)and α-Sb2O4:synthesis and characterization of some ketoximate modified antimony(III)alkoxides[J].Journal of SolGel Science and Technology.2010,54(1):119-128.
[9]KIM Dae-Wook,KIM Deok-Soo,KIM Young-Gon,et al.Preparation of hard agglomerates free and weakly agglomerated antimony doped tin oxide(ATO)nanoparticles by coprecipitation reaction in methanol reaction medium[J].Materials Chemistry and Physics.2006,97(2-3):452-457.
[10]毋伟,陈郑博,陈建峰.微乳液一步法制备纳米镍白油稳定分散体[J].稀有金属材料与工程.2009,38(S3):321-324.(WU Wei,CHEN Zheng-bo,CHEN Jian-feng.Preparation of Stable Dispersion of Nano-Nickel in White Oil by One-Step Method with a Microemulsion Reactor.Rare Metal Materials and Engineering[J].2009,38(3):321-324)
[11]BAKONYI Imre,TOTH-KADAR Eniko,POGANY Lajos,et al.Preparation and characterization of d.c.-plated nanocrystalline nickel electrodeposits[J].Surface and Coatings Technology.1996,78(1-3):124-136.
[12]XU Jianlin,CHEN Jidong,YANG Shuhua,et al.Effects of current density on copper nanoparticle prepared by electrochemical method[J].International Journal of Nanoparticles.2010,3(2):93 -103.
[13]CULLITY B.D..Elements of X-Ray Diffraction(M).2nd.USA:Addison-wesley publishing company,INC.1978,284.
[14]任俊,卢寿慈.固体颗粒的分散[J].中国粉体技术.1998,4(1):25-33.(Ren Jun,Lu Shouci.Dispersion of Solid Particles.Powder Science and Technology[J].1998,4(1):25 -33)
猜你喜欢
蓄电池(2022年1期)2022-02-25
建材发展导向(2021年15期)2021-11-05
食品安全导刊(2021年21期)2021-08-30
科学与财富(2021年33期)2021-05-10
能源工程(2021年1期)2021-04-13
汽车零部件(2018年5期)2018-06-13
科技创新与应用(2017年11期)2017-04-27
科技资讯(2015年8期)2015-07-02
建材发展导向(2014年2期)2014-05-04
食品工业科技(2014年21期)2014-03-11
