
浮石催化水中臭氧分解研究
2012-09-04 02:30:18沈吉敏陈忠林
哈尔滨工业大学学报 2012年10期
袁 磊,沈吉敏,陈忠林
(1.哈尔滨工业大学城市水资源与水环境国家重点实验室,150090哈尔滨;2.黑龙江省科学院自然与生态研究所,150040哈尔滨)
近年来,对臭氧氧化饮用水除污染技术的研究主要集中在提高臭氧在水中的传质效率和羟基自由基生成量与速率,强化臭氧对水中有机污染物的降解效果.非均相催化臭氧氧化作为一种具有应用前景的除污技术越来越受到重视[1-6].非均相催化剂通常为金属、金属氧化物和活性炭,因价格高、易被氧化等原因,限制其在大规模水处理中的应用,急需寻找可替代高效催化剂.国内外学者利用天然材料(砂、沸石、针铁矿、铝土矿)作为非均相催化剂,催化臭氧氧化水中有机物[7-10].浮石是一种多孔、轻质的玻璃质酸性火山喷出岩,储量丰富,其特点是质量轻、强度高、耐酸碱、抗腐蚀,且无污染、无放射性等,是理想的天然材料.目前,国内外关于浮石在水处理中的应用研究主要集中在吸附与光催化剂制备方面[11-14],未见浮石应用于臭氧体系的相关报道.本研究选用浮石作为催化剂,利用其高机械强度和化学稳定性,研究浮石催化水中臭氧分解途径,考察浮石表面化学性质对催化臭氧分解的影响,并探讨浮石催化水中臭氧分解机理.
1 实验
1.1 实验材料
浮石(五大连池,中国),粒径0.6~0.8mm,超声清洗两次,每次10min,用去离子水冲洗3遍,于60℃烘干备用.
1.2 实验方法
浮石催化臭氧分解实验在容量为1.2L的拟平底烧瓶反应器中进行(实验装置如图1所示).臭氧由DHX2SS21G型臭氧发生器(哈尔滨久久电化学工程技术有限公司,以氧气为气源,臭氧产量范围为0~9g/h)产生.通一定时间臭氧,使反应器中水溶液初始臭氧质量浓度恒定.用磁力搅拌器(300r/min)和恒温水浴装置(23℃)保持反应体系中臭氧质量浓度均匀和温度恒定.反应溶液的初始pH值(pH=6.86)用NaOH和HClO4溶液调节,催化剂投量为1g/L.
实验涉及的试剂均为分析纯或色谱纯,实验用水由Milli-poreMilli-Q纯水系统制备(电阻率≥18MΩ·cm).所用的容量玻璃仪器均用铬酸洗液浸泡,然后再用自来水和去离子水依次洗涤.非容量玻璃仪器在经上述处理后再经350℃高温处理过夜.

图1 实验工艺流程
1.3 分析方法
水溶液中臭氧质量浓度用靛蓝法测定[15],主要仪器为紫外-可见光分光光度计(T6,China).溶液pH值采用pH计(PB-10,Germany)测定.催化剂表面元素的化学组成采用X射线荧光光谱仪(PW4400,Holland)测定.催化剂表面性质采用低温氮吸附比表面和孔隙度测定仪(ASAP2020M,US)测定.采用热分析仪(STA-449C,Germany)研究催化剂的相结构与温度的关系.表面零点电荷(pHpzc)采用Zeta电位仪(ZetaSizer3000,UK)进行测定.表面羟基采用酸碱滴定法[16].
2 结果与讨论
2.1 浮石催化剂表面特征
浮石与其他用于非均相催化臭氧氧化的天然矿物相比,具有丰富蜂窝状孔结构、较大比表面积、高机械强度.本实验所用浮石经X射线荧光光谱仪(XRF)检测主要化学组成及其质量分数:SiO2(49.79%)、Al2O3(15.6%)、Fe2O3(9.15%)、K2O(5.31%)、MgO(4.15%)、Na2O(4.05%)和TiO2(3.13%),其表面性质测定结果见表1.

表1 浮石的表面性质
从图2可以看出,温度小于150℃时,浮石催化剂的失重很小,说明由于物理吸附而使表面结合的结晶水含量较小,在热重曲线上表现得不明显;当热处理温度升高至160~400℃时,热重曲线出现了大幅度的失重现象,质量损失约为1.13%.同时,在相应的温度区间,微商热重曲线有一个强度很大的吸热峰.此温度区间内的失重现象是因浮石的表面羟基受热后缩合失水所致.因此,随催化剂热处理温度升高,表面羟基密度降低.

图2 浮石的热重分析图
2.2 浮石催化水中臭氧分解的途径
臭氧在水中分解可表达为

则动力学方程为
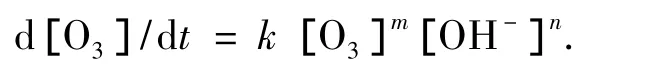
由于实验中pH值不变,[OH-]可看作常数,其动力学方程可简化为
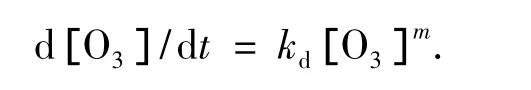
式中kd=[OH-]n,因此,由臭氧质量浓度随时间变化可求出kd(表观速率常数或臭氧分解速率常数)及相对于臭氧的反应级数m.
实验考察了温度23℃,初始pH值6.86条件下,水中臭氧分解和浮石催化臭氧分解效果,并进行ln([O3]t/[O3]0)-t的拟合,其中[O3]t为反应时间t时水样中臭氧的瞬时质量浓度(mg/L),[O3]0为原水中臭氧的初始质量浓度0.6mg/L,t为反应时间(min),拟合结果如图3所示.臭氧在水中分解和浮石催化水中臭氧分解都较好地符合一级反应.浮石催化臭氧分解的一级分解速率常数提高了37.4%,显著促进水中臭氧分解.加速水中臭氧分解可能有3种途径:浮石有吸附水中臭氧的作用;臭氧氧化浮石的还原性表面;浮石催化臭氧分解生成自由基.

图3 不同工艺水中臭氧的ln([O3]t/[O3]0)-t拟合图
水中臭氧的分解可用以下机理反应方程式表示[17]:




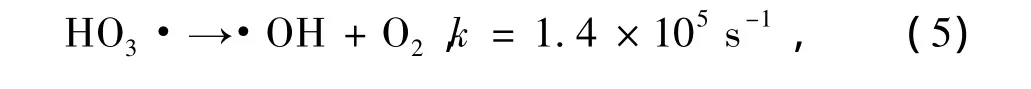

反应式(1)是臭氧在水中分解的限速步骤,每3分子的臭氧分解可以生成两分子的羟基自由基.由反应式(6)可以看出,羟基自由基和水中臭氧分子的反应速度非常快.由于去离子水中可消耗羟基自由基的物质的量非常少,水中臭氧的消耗主要来源于臭氧分解生成的羟基自由基,如果能抑制这种消耗就能显著降低臭氧在去离子水中的分解速度.同理,如果引入羟基自由基捕获剂使催化臭氧在水中分解的速度明显降低,则表明催化过程生成了羟基自由基(·OH).
叔丁醇是一种典型的·OH淬灭剂[18],它与·OH的反应速率常数为5×108(mol/L)-1·s-1,在溶液中可以快速地与·OH发生反应,并且反应后不产生·HO2和·O-2,相当于臭氧分解链反应的终止剂,而与臭氧基本不发生反应.因此,通过考察叔丁醇的影响可以间接地判断反应是否遵循·OH机理.
实验考察了23℃、初始pH值6.86,叔丁醇质量浓度为10mg/L,原水中臭氧的初始质量浓度0.6mg/L条件下,水中臭氧分解和浮石催化水中臭氧分解效果,并进行ln([O3]t/[O3]0)-t的拟合,结果如图4所示.由图3、4可以看出,加入叔丁醇后水中臭氧分解和浮石催化臭氧分解速率常数分别降低29%和45.2%,对浮石催化水中臭氧分解抑制效果尤为明显.因此,根据实验数据得出结论,浮石促进水中臭氧分解,并在分解过程中生成了羟基自由基.
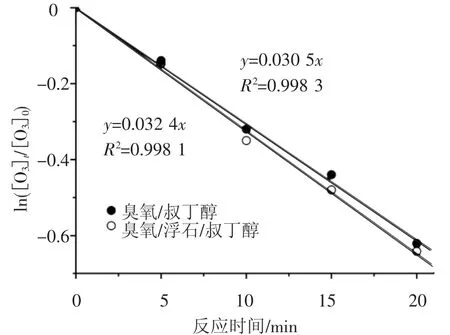
图4 叔丁醇存在水中臭氧的ln([O3]t/[O3]0)-t拟合图
2.3 热处理温度对臭氧分解的影响
考察了23℃、反应20 min,催化水中臭氧分解效果,其中原水中臭氧的初始质量浓度为0.6 mg/L,结果如图5所示.可以看出,浮石催化臭氧分解,随浮石催化剂处理温度的升高,水中臭氧分解率降低.浮石热处理温度由60℃提高到400℃,催化臭氧分解速率常数由0.059 2减少到0.045 3,降低了23.5%.
非均相催化臭氧化工艺中催化剂催化水中臭氧分解的能力与其表面性质直接相关[19].表面羟基是与臭氧分子直接接触的表面物种,因此,了解表面羟基的密度和状态有助于理解催化剂催化臭氧分解的活性.由图2可以看出,随催化剂热处理温度的升高,催化剂表面羟基缩合失水.温度由60℃提高到400℃,浮石表面羟基密度由0.27 mmol/g减少到0.05 mmol/g,降低81.4%.因此,随浮石催化剂热处理温度升高,催化水中臭氧分解能力降低,是由于浮石表面羟基缩合、失去表面羟基所致.浮石催化剂表面羟基密度与水中臭氧分解效率成正相关.因此,可以推断浮石催化水中臭氧分解起主要作用的是催化剂的表面羟基.

图5 热处理温度对臭氧分解的影响
2.4 溶液pH值对催化臭氧分解的影响
溶液pH值是影响水中臭氧分解的重要因素,臭氧分解程度与 OH-质量浓度关系密切,OH-质量浓度越大,臭氧越容易进行分解反应产生·OH(反应式(1)~(6)).为了研究表面电荷状态对催化水中臭氧分解的影响,以浮石为催化剂考察其在不同pH条件下的催化活性,结果如图6.实验考察了23℃、单独臭氧分解和浮石催化水中臭氧分解效果,其中原水中臭氧初始质量浓度为0.6 mg/L.可以看出,单独臭氧和浮石催化臭氧分解率随pH值增大而提高.当溶液pH值在5.01~6.86时浮石表现出较强臭氧分解能力.溶液中催化剂通过水-催化剂界面接触水中的臭氧分子,水中催化剂催化臭氧分解的能力与其表面性质有直接关系.表面零质子电荷点pHpzc(point of zero charge)是指催化剂表面电荷为零时的pH值[20],与催化剂本身的结构组成有关.当溶液pH值接近催化剂的pHpzc时,催化剂表面基本是中性状态,即此时由质子平衡达到表面正负电荷总量基本相等.若溶液的pH值与催化剂的pHpzc有所差别,催化剂在水中表面电荷状态会发生变化,呈现质子化(pH <pHpzc)或去质子化(pH>pHpzc)状态,即

溶液pH≈6.22(pHpzc)时,浮石具有较强的催化分解水中臭氧能力.溶液pH=3.01时,浮石没有表现出较强催化活性,这可能是因为在酸性条件下浮石表面被较强质子化,由于臭氧分子的亲电特性,不易吸附到催化剂表面,抑制浮石参与催化反应.而当溶液pH=8.93时,溶液中OH-与浮石产生竞争反应,而OH-与臭氧结合促使其分解产生自由基的概率和能力远大于催化剂表面对臭氧的吸附催化分解,所以,催化活性也受到一定抑制,但溶液中OH-增加,臭氧分解速度相对加快.因此,可以肯定只有—OH状态的表面可以引发臭氧生成羟基自由基,—O-和—OH2+状态的表面都没有这种活性.

图6 溶液pH值对催化臭氧分解的影响
2.5 催化臭氧分解引发·OH机理的探讨
XRF分析表明,实验用浮石催化剂的主要化学成分为二氧化硅和金属氧化物.由此,推测促进水中臭氧分解的是浮石表面金属氧化物的表面羟基.金属氧化物中的表面羟基可以分为两类:一类是催化剂本身结构中的表面羟基;另一类是由于水合作用在催化剂表面形成的一层羟基.酸碱滴定测得浮石表面羟基密度为0.27 mmol/g.将浮石作为催化剂放入溶液中进行催化臭氧化反应时,浮石表面羟基成为臭氧分解的活化中心,由于臭氧自身结构特点与催化剂表面羟基形成配位键和氢键,形成五元环结构.催化剂表面羟基作用于此时臭氧分子,共价键扭曲松弛,分解生成氧分子,同时在浮石表面形成活性物质—HO2-,—HO2-再与臭氧分子结合生成O3-·和 HO3·,HO3·进一步分解生成HO·;催化剂表面的—O2-·结合臭氧产生O3-·和O2,同时发生脱附,表面形成空穴重新结合水分子生成—H2O,O3-·和—H2O反应生成HO3·,催化剂表面重新形成羟基,完成一个反应循环.在此反应链中有3分子臭氧参与反应生成两分子羟基自由基,分解生成的羟基自由基从催化剂表面扩散,完成在固液界面分解臭氧生成羟基自由基,并向溶液体系氧化传递过程.分散到溶液中的高氧化性羟基自由基进而氧化水中污染物.
3 结论
1)浮石能显著提高水中臭氧的分解速率,并生成羟基自由基.
2)浮石表面金属氧化物羟基是分解水中臭氧的活性中心,表面羟基密度与水中臭氧分解率成正相关.
3)随溶液pH值升高,浮石催化水中臭氧分解率升高,浮石的零电荷pH值(pHpzc)决定了特定水溶液pH值时的表面电荷状态,当水溶液中的浮石表面接近零电荷时,浮石催化水中臭氧分解活性最强.
[1]XU Zhenzhen,CHEN Zhonglin,JOLL C,et al.Catalytic efficiency and stability of cobalt hydroxide for decomposition of ozone and p-Chloronitrobenzene in water[J].Catal Commun,2009,10(8):1221-1225.
[2]YANG Yixin,MA Jun,QIN Qingdong,et al.Degradation of nitrobenzene by nano-TiO2catalyzed ozonation[J].J Mol Catal,A,2007,267(1/2):41 -48.
[3]MA Jun,SUI Minghao,CHEN Zhonglin,et al.Degradation of refractory organic pollutants by catalytic ozonation-activated carbon and Mn-loaded activated carbon as catalysts[J].Ozone Sci Eng,2004,26(1):3 - 10.
[4] MA Jun,SUI Minghao,ZHANG Tao,et al.Effect of pH on MnOx/GAC catalyzed ozonation for degradation of nitrobenzene[J].Water Res,2005,39(5):779 -786.
[5]SUN Zhizhong,MA Jun,WANG Libo,et al.Degradation of nitrobenzene in aqueous solution by ozone-ceramic honeycomb[J].J Environ Sci,2005,17:716 -721.
[6]ZHAO Lei,MA Jun,SUN Zhizhong.Oxidation products and pathway of ceramic honeycomb-catalyzed ozonation for the degradation of nitrobenzene in aqueous solution[J].Appl Catal,B,2008,79(3):244 -253.
[7]CHOI H,KIM Y,LIM H,et al.Oxidation of polycyclic aromatic hydrocarbons by ozone in presence of sand[J].Water Sci Technol,2001,43:349 -356.
[8]FUJITA H,IZUMI J,SAGEHASHI M,et al.Adsorption and decomposition of water-dissolved ozone on high silica zeolites[J].Water Res,2004,38(1):159-165.
[9]PARK J,CHOI H,AHN K,et al.Removal mechanism of natural organic matter and organic acid by ozone in the presence of goethite[J].Ozone Sci Eng,2004,26(2):141-152.
[10]QI Fei,XU Bingbing,CHEN Zhonglin,et al.Ozonation catalyzed by the raw bauxite for the degradation of 2,4,6-trichloroanisole in drinking water[J].J Hazard Mater,2009,168(1):246 -252.
[11]BEKAROGLU S S K,YIGIT N O,KARANFIL T,et al.The adsorptive removal of disinfection by-product precursors in a high-SUVA water using iron oxide-coated pumice and volcanic slag particles[J].J Hazard Mater,2010,183(1/2/3):389-394.
[12]YAVUZ M,GODE F,PEHLIVAN E,et al.An economic removal of Cu2+and Cr3+on the new adsorbents:pumice and polyacrylonitrile/pumice composite[J].Chem Eng J,2008,137(3):453 -461.
[13]RAO K V S,SUBRAHMANYAM M,BOULE P.Immobilized TiO2photocatalyst during long-term use:decrease of its activity[J].Appl Catal,B,2004,49(4):239-249.
[14]RAO KVS,RACHEL A,SUBRAHMANYAM M,et al.Immobilization of TiO2on pumice stone for the photocatalytic degradation of dyes and dye industry pollutants[J].Appl Catal,B,2003,46(1):77 -85.
[15]BADER H,HOIGNE J.Determination of ozone in water by the indigo method[J].Water Res,1981,15(4):449-456.
[16]TAMURA H,TANAKA A,MITA K,et al.Surface hydroxyl site densities on metal oxides as a measure for the ion-exchange capacity[J].J Colloid Interface Sci,1999,209(1):225-231.
[17]GUNTEN V U.Ozonation of drinking water:oxidation kinetics and product formation[J].Water Res,2003,37(7):1443-1467.
[18]LANGLAIS B,RECKHOW D A,DEBORAH R B.O-zone in water treatment:application and engineering[M].Michigan:Lewis Publishers,1991:18-19.
[19]马军,张涛,陈忠林,等.水中羟基化铁催化臭氧分解和氧化痕量硝基苯的机理探讨[J].环境科学,2005,26(2):78-82.
[20]MULLET M,FIEVET P,SZYMCZYK A,et al.A simple and accurate determination of the point of zero charge of ceramic membranes[J].Desalination,1999,121(1):41-48.
猜你喜欢
煤气与热力(2021年10期)2021-12-02 05:11:46
文萃报·周五版(2020年22期)2020-06-08 00:10:17
中成药(2018年2期)2018-05-09 07:20:05
——《青瓷》作者的人生哲学
爱你(2017年35期)2017-12-08 07:47:07
—— 《青瓷》作者的人生哲学
爱你(2017年32期)2017-11-10 02:56:39
中学生数理化·八年级物理人教版(2017年12期)2017-04-18 12:59:46
中国蔬菜(2016年8期)2017-01-15 14:23:43
新乡学院学报(2016年6期)2016-12-01 05:21:38
当代化工研究(2016年9期)2016-03-20 16:22:11
少儿科学周刊·少年版(2015年1期)2015-07-07 21:57:30
