
HPLC 法测定伏立康唑乳剂的含量及其有关物质
2012-08-06 09:52张新事付淑军林小燕李玉娟北京理工大学生命学院北京100081国际生物医药联合研究院瀚盟生物技术天津有限公司天津300457
中国药房 2012年1期
张新事,付淑军,林小燕,李玉娟(1.北京理工大学生命学院,北京100081;.国际生物医药联合研究院/瀚盟生物技术(天津)有限公司,天津 300457)
伏立康唑(Voriconazole)为美国辉瑞公司开发的第2代三唑类抗真菌药物,2002年8月在美国首次上市。伏立康唑口服和静脉给药均有很好的抗真菌活性,对细菌、霉菌和酵母菌均有强抑菌作用,是治疗深部真菌感染首选药[1~4]。目前临床上主要使用的是伏立康唑静脉输注用冻干粉末。由于伏立康唑水溶性很差,因此该制剂中加入了大量硫代丁基醚-β-环糊精进行增溶,但也导致可能存在给药时刺激性强、易发生溶血等不良反应[5]。北京理工大学生命学院生物分析实验室采用独特的乳化技术,利用伏立康唑可在油水两相中形成非均匀的水包油型液体分散体系的特点,将其制成静脉注射用乳剂。由于不含大量的增溶剂,可减少由此而导致的溶血等不良反应,还可降低给药时产生的刺激性,减少药物的毒副作用。
鉴于伏立康唑乳剂基质的复杂性,干扰成分多,因此有必要建立灵敏、专属的方法进行主药及其有关物质含量测定。本文采用高效液相色谱(HPLC)法,建立了伏立康唑乳剂的含量测定方法。结果表明,该方法简便、准确,专属性强,可用于伏立康唑乳剂的含量测定及其有关物质的控制。
1 仪器与试药
HPLC系统,包括LC-10AT输液泵、SPD-10A紫外检测器(日本岛津公司);N-2000色谱工作站(浙江大学);乙腈、甲醇均为色谱纯,其余试剂均为分析纯。
伏立康唑对照品(万特制药(海南)有限公司,批号:20100303,纯度:99.9%);伏立康唑乳剂(北京理工大学生命学院生物分析实验室自制,批号:100701、100702、100703,规格:100 mg∶25 mL)。
2 方法与结果
2.1 色谱条件及系统适用性试验
色谱柱:Diamonsil C18(250 mm×4.6 mm,5 μm);流动相:乙腈-水-乙酸(40∶60∶0.25,V/V/V),流速:1.0 mL·min-1;检测波长:256 nm;灵敏度:0.01 AUFS;柱温:室温;进样量:20 μL。理论板数按伏立康唑峰计应不低于1 500。
2.2 溶液的制备
2.2.1 空白基质溶液:精密量取本品的空白基质0.1 mL(按照乳剂处方所用赋形剂的种类和用量制备,但不含伏立康唑),置于25 mL容量瓶中,加无水乙醇5 mL后振摇,再以无水乙醇定容至刻度,摇匀,即得。
2.2.2 对照品溶液:精密称定减压干燥至恒重的伏立康唑对照品1.00 mg,加无水乙醇溶解并逐级稀释成浓度为100.0、50.0、20.0、10.0、5.00、2.00、1.00 μg·mL-1的溶液,即得。
2.2.3 供试品溶液:精密量取本品0.1 mL(相当于伏立康唑0.4 mg),置于25 mL容量瓶中,加无水乙醇5 mL后振摇,再以无水乙醇定容至刻度,摇匀,即得。
2.2.4 样品破坏溶液:精密量取本品0.1 mL(相当于伏立康唑0.4 mg)5份,各置于25 mL容量瓶中,其中3份分别加入0.1 mol·L-1氢氧化钠、0.1 mol·L-1盐酸、3%双氧水各0.2 mL,室温放置2 h,加无水乙醇5 mL后振摇,再以无水乙醇定容至刻度,摇匀,即得碱、酸和氧化破坏后样品溶液;另2份置于25 mL容量瓶中,分别于4 500 lx条件室温下放置5 d及(40±5)℃下放置5 d,加无水乙醇5 mL后振摇,再以无水乙醇定容至刻度,摇匀,即得光照破坏溶液和高温破坏样品溶液。
2.3 专属性试验
取空白溶剂(无水乙醇)、空白基质、对照品和供试品溶液进样分析,色谱见图1;各破坏后样品溶液色谱见图2。
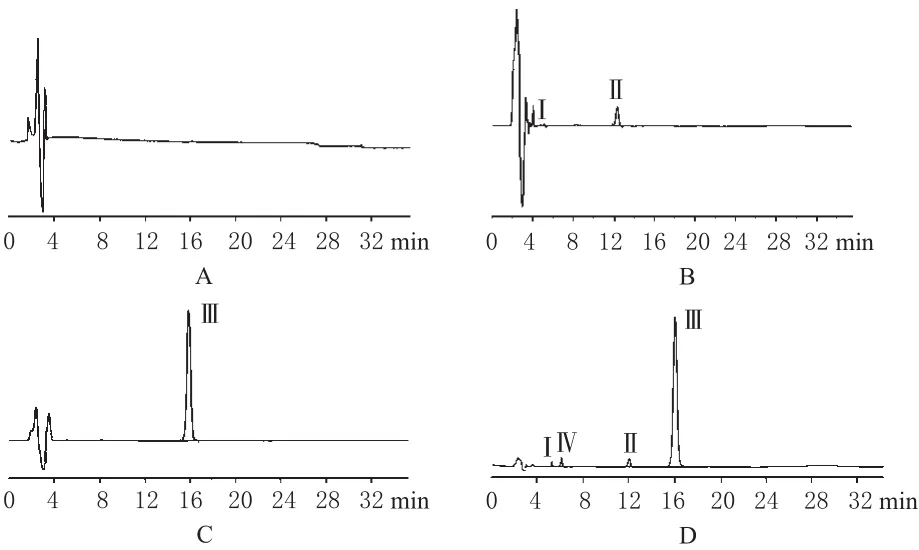
图1 高效液相色谱图A.空白溶剂;B.空白基质;C.对照品;D.供试品;Ⅰ、Ⅱ.来自赋形剂的干扰峰;Ⅲ.伏立康唑;Ⅳ.可能的降解产物Fig 1 HPLC chromatogramsA.blank solvent;B.blank matrix;C.substance control;D.test sample;Ⅰ,Ⅱ:interfering peaks from excipients;Ⅲ:voriconazole;Ⅳ:potential degradation product
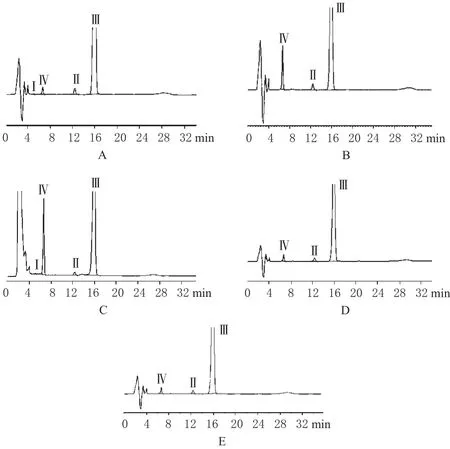
图2 破坏样品溶液的高效液相色谱图A.酸破坏样品溶液;B.碱破坏样品溶液;C.光照破坏样品溶液;D.高温破坏样品溶液;E.氧化破坏样品溶液;Ⅰ、Ⅱ.来自赋形剂的干扰峰;Ⅲ.伏立康唑;Ⅳ.可能的降解产物Fig 2 HPLC chromatograms of destroyed sample solution A.solution destroyed by acid;B.solution destroyed by alkal;C.solution destroyed by light;D.solution destroyed by heat;E.solution destroyed by oxidation;Ⅰ,Ⅱ.interfering peaks from excipients;Ⅲ.voriconazole;Ⅳ.potential degradation product
由图1可见,在空白溶剂的色谱图(图1A)中未发现有干扰峰的存在;而在空白基质的色谱图(图1B)中含有2个杂质峰,其保留时间分别为5.2、6.0、12.4 min(注:3个化合物尚未鉴定);在对照品的色谱图(图1C)中,伏立康唑的保留时间为15.9 min。表明溶剂及乳剂中的赋形剂均不干扰伏立康唑的测定。
由图2可见,伏立康唑对照品溶液经碱破坏后出现保留时间为6.7 min的色谱峰,另外本品在酸、氧化、光照及高温条件下均出现保留时间为6.7 min的色谱峰,尤其在碱、氧化和高温破坏样品中含量较高,因此可确定其为伏立康唑乳剂中的有关物质。比较上述色谱图可知,乳剂中赋形剂、降解产物、伏立康唑以及溶剂色谱峰彼此之间均分离良好。因此本方法对伏立康唑乳剂中伏立康唑的测定具有良好的专属性。
2.4 线性关系考察
精密称定减压干燥至恒重的伏立康唑对照品1.00 mg,加无水乙醇溶解并逐级稀释成浓度为100.0、50.0、20.0、10.0、5.00、2.00、1.00 μg·mL-1的溶液,各取20 μL,依次注入液相色谱仪,记录色谱。以伏立康唑的色谱峰面积(y)对浓度(x)回归,计算线性回归方程为y=2×106x+5 735.5(r=0.999 9),结果表明伏立康唑检测浓度线性范围为1.00~100.0 μg·mL-1。
2.5 检测限与定量限试验
称取伏立康唑对照品适量,以无水乙醇溶解并稀释到一定浓度,吸取20 μL注入液相色谱仪,记录色谱。分别以伏立康唑色谱峰响应值约为基线噪音的3倍(信噪比为3)和10倍(信噪比为10)时的进样浓度为检测限和定量限。结果,检测限为0.3 ng·mL-1,定量限为1.0 ng·mL-1。
2.6 稳定性试验
按“2.2.2”项下方法制备供试品溶液,制得后即放置在室温条件下,分别于第0、2、5、10、25 h取样20 μL,注入液相色谱仪,记录色谱峰面积,计算其RSD=0.71%。表明供试品溶液室温放置25 h内性质稳定。
2.7 重复性试验
取同一批号的样品,制备成供试品溶液,连续进样测定6次,结果RSD=0.33%,表明本方法重复性良好。
2.8 中间精密度试验
取同一批号的样品,由2名分析者,分别在2 d内在2台不同的色谱仪上进行含量测定,考察中间精密度,结果RSD=1.80%,表明本方法中间精密度良好。
2.9 回收率试验
平行量取已知含量的乳剂9份,每份0.10 mL,分别置于25 mL容量瓶中,依次加入低、中、高3种浓度的对照品溶液5.0 mL,制备供试品溶液,分别取20 μL注入液相色谱仪,记录色谱图,并按外标法以峰面积计算加样回收率。结果,低、中、高3个浓度的平均加样回收率为98.8%,RSD<2%,表明本方法具有良好的准确度,详见表1。
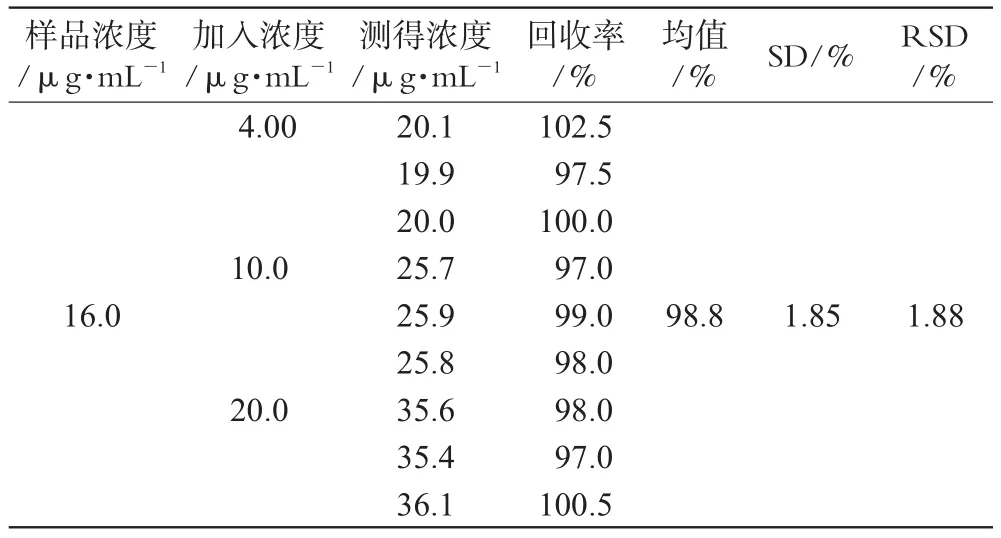
表1 回收率试验结果Tab 1 Results of recovery test
2.10 样品中主药含量测定
精密量取本品0.10 mL(相当于伏立康唑400 μg),置于25 mL容量瓶中,加无水乙醇5.0 mL,振摇,继续以无水乙醇稀释到刻度,摇匀,以0.45 μm微孔滤膜过滤,即得供试品溶液。取上述供试品溶液20 μL注入色谱仪,记录色谱图,按外标法以峰面积计算含量。结果,3批伏立康唑乳剂的含量测定结果均符合规定,详见表2。

表2 3批样品含量测定结果Tab 2 Results of content determination of 3 batches of samples
2.11 样品中有关物质含量测定
量取本品1.0 mL,置于25 mL容量瓶中,加无水乙醇5.0 mL,振摇,以无水乙醇稀释到刻度,摇匀;以0.45 μm微孔滤膜过滤,即得约含160 μg·mL-1伏立康唑的供试品溶液。另精密量取适量对照品贮备液,以无水乙醇稀释制成约含1.6 μg·mL-1伏立康唑的对照溶液。照“2.1”项下的色谱条件,量取对照溶液20 μL,注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的10%~20%;精密量取供试品溶液和对照溶液各20 μL,分别注入液相色谱仪,记录色谱图至主成分峰保留时间的2倍。供试品溶液色谱图中如有杂质峰,各杂质的峰面积之和不得大于对照液中主成分峰面积的3倍(3.0%)。经检测,3批伏立康唑乳剂的有关物质含量分别为1.13%、1.03%和1.02%。
3 讨论
在前期试验中,笔者分别考察了2种比例流动相系统乙腈-水-乙酸(55∶45∶0.25,40∶60∶0.25,V/V/V)对分析的影响。当以前者为流动相时,伏立康唑的保留时间为7.0 min,分析时间适中。但在进行相关物质的影响因素试验时,发现分离度较差,峰形不对称,因此继续对流动相的比例进行了优化,最后确定后者为流动相。在此条件下,伏立康唑与杂质能够完全分开,峰形对称,伏立康唑的保留时间为15.9 min。在上述色谱条件下,主要存在3个可能的杂质峰,其与伏立康唑以及彼此之间的分离度均大于1.5,符合有关要求。
在伏立康唑乳剂中,伏立康唑被包合于水包油的乳粒中,若要对其进行含量测定,首先应采用适当的有机溶剂实施破乳,释放伏立康唑。本试验考察了用于破乳的多种有机溶剂,包括甲醇、无水乙醇、乙腈等,结果发现无水乙醇的破乳效果最好,并且适用于随后的HPLC分析。
伏立康唑乳剂中含有精制蛋黄磷脂、油酸、注射用大豆油、甘油、维生素E等赋形剂,基质组成相对复杂,可能给含量及其有关物质测定带来干扰。文献[4]报道方法均为原料药或注射剂的分析检测方法,与乳剂相比,其基质相对简单,干扰较少;而本实验室制备的乳剂基质复杂,采用文献报道方法干扰多,不适用于本研究。因此,本试验成功建立了用于伏立康唑乳剂的主药及有关物质含量测定的HPLC法,经方法验证,确认本方法准确可靠、专属性强,适用于伏立康唑乳剂的快速分析。
[1]尚茂林.抗真菌新药——伏立康唑[J].中国药房,2007,18(19):1 505.
[2]张志华,易 鸿,何周康,等.HPLC-MS/MS法测定伏立康唑的血药浓及其在生物等效性研究中的应用[J].中南药学,2009,7(12):889.
[3]昌 盛.伏立康唑[J].中国药物化学杂志,2007,17(3):197.
[4]吴良法.反相高效液相色谱法测定伏立康唑的含量[J].中国药房,2007,18(1):58.
[5]官东秀,冯祚臻,俸小平,等.伏立康唑药物不良反应文献分析[J].齐鲁药事,2009,28(8):507.
猜你喜欢
发明与创新(2022年31期)2022-11-03
新型工业化(2022年3期)2022-06-18
中国医药科学(2022年5期)2022-05-05
广州化工(2022年6期)2022-04-11
安徽农学通报(2022年6期)2022-04-07
药学服务与研究(2020年4期)2020-09-03
化工设计通讯(2020年1期)2020-03-04
中国抗生素杂志(2019年7期)2019-07-25
中国医院用药评价与分析(2018年10期)2018-11-12
中国感染与化疗杂志(2015年4期)2015-01-24
