
HPLC法同时测定缬沙坦氢氯噻嗪胶囊中缬沙坦和氢氯噻嗪的含量
2012-03-26 05:36:10吴迪郭伟英辽宁医学院辽宁锦州121001
中国药房 2012年9期
吴迪,郭伟英(辽宁医学院,辽宁锦州121001)
缬沙坦氢氯噻嗪胶囊是由缬沙坦和氢氯噻嗪组成的复方制剂,用于治疗单一药物不能充分控制血压的轻-中度原发性高血压。缬沙坦作为一种血管紧张素Ⅱ(AngⅡ)受体拮抗药,是90年代出现的新型抗高血压药物。缬沙坦安全而疗效肯定,优于钙拮抗药、噻嗪类利尿剂和血管紧张素转换酶抑制剂[1]。但当缬沙坦的使用剂量增加到一定水平后,其药效并不随剂量增强,多数患者的治疗需与其他降压药联合应用才能达到有效降压目的[2],因此,缬沙坦氢氯噻嗪胶囊制剂的出现是解决以上问题的较好方案[3,4]。
噻嗪类利尿剂主要作用部位为髓袢升支皮质部及远曲小管起始部,短期使用时主要通过抑制钠离子、氯离子和水的重吸收,达到排钠利尿作用,使血容量和细胞外液减少、心输出量下降,从而达到降压作用。而长期使用利尿剂后血容量不会进一步下降,这时噻嗪类利尿剂可通过直接舒张血管、降低血管顺应性而使血压降低[5]。此外,单用氢氯噻嗪还可能引发低血钾症,当其与缬沙坦联合应用时则可以减少低血钾的发生,原因是缬沙坦既可以减少利尿剂的用量又能减轻因利尿剂引起的电解质变化[2]。因此,相关文献中专家建议沙坦类降压药物应与利尿剂同时使用。
缬沙坦单方制剂[6]和氢氯噻嗪单成分含量测定方法也已有报道[7],但是报道中方法大多操作烦琐、误差范围大、流动相中含有缓冲盐溶液,这样不利于试验的简便操作或不利于色谱柱的保护,本课题采用的高效液相色谱-紫外(HPLC-UV)法只要求样品制成溶液,且选用制备简单、含酸的流动相,对色谱柱损伤相对较小,同时还具有分辨率高、灵敏度高、速度快等优点,能在同一种条件下同时测定出2种成分的含量,既节省时间又可以保证测定的准确度和精密度等要求,所以本文采用此法测定该制剂中缬沙坦和氢氯噻嗪的含量。
1 仪器与试药
FL2200液相色谱仪、FL2200 UV检测器、FL9500色谱工作站(浙江温岭福立分析仪器有限公司);KQ-100A型超声清洗器(昆山市超声仪器有限公司);TD型电子天平(德国赛多利斯公司)。
缬沙坦标准品(批号:100651-200902,纯度:98.9%)、氢氯噻嗪标准品(批号:100309-200702,纯度:99.8%)均由中国食品药品检定研究院提供;缬沙坦氢氯噻嗪胶囊(山东鲁抗辰欣药业有限公司,批号:100324201、100328201、100528204,每粒胶囊含缬沙坦80 mg、氢氯噻嗪12.5 mg);甲醇、磷酸均为色谱纯,其余试剂均为分析纯,水为超纯水。
2 方法与结果
2.1 溶液制备
2.1.1 混合标准品溶液的制备。精密称取缬沙坦和氢氯噻嗪标准品80、12.5 mg,置于同一100 mL量瓶中,加流动相超声溶解并稀释至刻度,摇匀,得标准品贮备液。精密吸取此贮备液10 mL置于100 mL量瓶中,加流动相稀释至刻度,摇匀,即得。
2.1.2 供试品溶液的制备。取缬沙坦氢氯噻嗪胶囊10粒,去除胶囊壳精密称取内容物适量(约相当于缬沙坦80 mg),置于100 mL量瓶中,加流动相适量,超声溶解,放冷,用流动相定容,摇匀,滤过。精密量取续滤液10 mL,置于100 mL量瓶中,加流动相定容,摇匀,静置,吸取上清液,经0.45 μm微孔滤膜过滤,即得供试品溶液。
2.1.3 阴性样品液的制备。取处方组成中除缬沙坦和氢氯噻嗪外的其他成分,制成阴性样品,按供试品溶液的制备方法处理得阴性样品液。
2.2 色谱条件与系统适用性试验
色谱柱:Chromatrex C18(200 mm×4.6 mm,5 μm);流动相:甲醇-水(70∶30,用磷酸调pH至3.1),流速:1.0 mL·min-1;检测波长:230 nm;柱温:25℃;进样量:20 μL。理论板数按缬沙坦和氢氯噻嗪计算均在4 000以上,分离度均>1.5,阴性样品液不干扰测定。标准品、供试品和阴性样品色谱见图1。

图1 高效液相色谱图Fig 1 HPLC chromatograms
由图1表明,在本试验色谱条件下,样品中其他成分对缬沙坦和氢氯噻嗪的测定无干扰,氢氯噻嗪和缬沙坦保留时间分别约为2.6、7.9 min。
2.3 线性关系考察
分别精密量取标准品贮备液1、2、5、10、15、20 mL,置于100 mL量瓶中,加流动相溶解定容,摇匀,即得系列溶液。分别取上述溶液进样测定,以峰面积(A)为纵坐标,进样浓度(c)为横坐标,进行线性回归,得缬沙坦和氢氯噻嗪回归方程分别为A=4.723 6×104c+4.008 6×105(r=0.999 7)、A=7.024 4×104c+2.790 4×104(r=0.999 5)。结果表明,缬沙坦和氢氯噻嗪检测浓度线性范围分别为8~160、1.25~25 μg·mL-1。
2.4 精密度试验
取混合标准品溶液,重复进样6次,得缬沙坦和氢氯噻嗪峰面积的RSD分别为0.35%、0.13%。
2.5 重复性试验
取同批样品,共6份,按“2.1.2”项下方法制备供试品溶液,进样测定,结果,缬沙坦和氢氯噻嗪含量的RSD分别为1.6%和1.1%。
2.6 稳定性试验
取供试品溶液,4 ℃避光放置,于0、1、2、4、8 h分别进样20 μL。结果,缬沙坦和氢氯噻嗪峰面积的RSD分别为0.76%和0.48%,表明供试品溶液在8 h内基本稳定。
2.7 回收率试验
精密称取同批已知含量的缬沙坦氢氯噻嗪胶囊的内容物,共9份,平均分为3组,分别按80%、100%、120%的量加入标准品,按“2.1.2”项下方法制备供试品溶液,进样测定。按外标法计算缬沙坦、氢氯噻嗪的平均回收率,结果见表1。
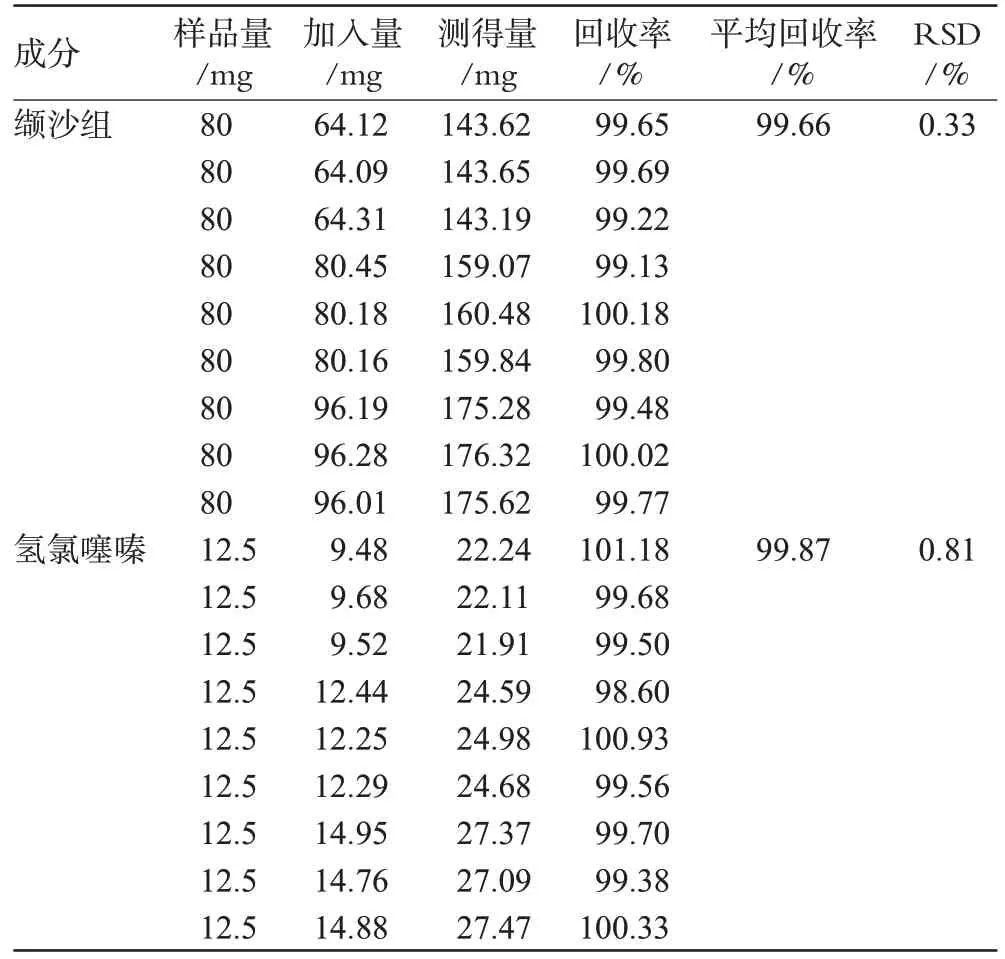
表1 回收率试验结果(n=9)Tab 1 Results of recovery test(n=9)
2.8 样品含量测定
分别精密称取不同批号的样品,按“2.1.2”项下方法制备供试品溶液,进样测定,记录色谱峰,按外标法以峰面积计算缬沙坦、氢氯噻嗪含量(占标示量),结果见表2。

表2 样品含量测定结果(n=3)Tab 2 Results of content determination of samples(n=3)
3 讨论
3.1 提取条件的选择
本品含量测定所用溶剂为流动相,因缬沙坦在此流动相中易溶;而氢氯噻嗪需超声溶解,因此在含量测定中,制备标准品溶液与供试品溶液时均需要进行超声处理。
3.2 检测波长的选择
本方法采用在230 nm波长处同时测定缬沙坦和氢氯噻嗪2种成分的含量,因为本品为复方制剂,同时测定两者的含量可使质控方法更简便。缬沙坦在230 nm波长附近有最大吸收,而氢氯噻嗪在较长波长范围内(200~400 nm)都有较大的吸收,在230 nm波长处吸收亦较大,故采用230 nm作为二者的检测波长。
3.3 流动相的选择
笔者曾选用甲醇-水及乙腈-水为流动相,两者分离度相近,但是采用前者时氢氯噻嗪拖尾因子比乙腈大,而当调整甲醇和水的比例并调节pH值后,既避免了氢氯噻嗪峰的拖尾现象,又使得样品中各杂质峰能与缬沙坦、氢氯噻嗪峰有效分离。此外,色谱级的乙腈价格比色谱级甲醇的价格高很多,且毒性大。因此在保证分离效果的前提下,最终选定甲醇-水的流动相组成(70∶30,用磷酸调pH为3.1)用于本次分析。
3.4 方法的确定
目前国内生产此药的生产厂家很少,对此药的研究也非常少。国内未见有关缬沙坦氢氯噻嗪胶囊中氢氯噻嗪含量测定方法的文献报道;有测定复方缬沙坦片中缬沙坦含量的文献报道[8],但是此方法不能很好地分离缬沙坦和氢氯噻嗪。本研究建立的HPLC-UV法能在同一种条件下同时测定出2种成分含量,既节省时间又可以保证测定的准确度和精密度,而且峰形良好,分离度、拖尾因子等也达到了要求。因此,本研究为缬沙坦氢氯噻嗪胶囊的质量控制提供了一种简便且省时的方法。
国外通常采用HPLC-二极管阵列检测器(DAD)法同时测定2种成分的含量[9],由于缬沙坦氢氯噻嗪胶囊中缬沙坦和氢氯噻嗪的含量相差较大,对检测器的灵敏度要求较高,DAD检测器的灵敏度比UV检测器要低,所以,本研究建立的HPLCUV法灵敏度更高。
[1]范治国,刘治安.抗高血压药的研究进展与临床应用评价[J].中国药房,2010,21(42):4 011.
[2]孙宁玲.血管紧张素受体拮抗剂联合利尿剂的治疗方案在高血压治疗中的地位[J].中华高血压杂志,2010,18(8):714.
[3]赵秀丽,胡大一.血管紧张素Ⅱ受体拮抗剂-缬沙坦研究进展[J].中国心血管杂志,1998,3(3):218.
[4]陈秋琴,吴慧英,翁春梅,等.缬沙坦的临床应用进展[J].中国基层医药,2005,12(5):627.
[5]Ernst ME,Moser M.Use of diuretics in patients with hypertension[J].N Eng J Med,2009,361(22):2 153.
[6]廖红娟,张 涛.高效液相色谱法测定缬沙坦片的含量[J].中国药业,2010,19(9):36.
[7]饶五湖,林瑞群.高效液相色谱法测定氢氯噻嗪片的含量[J].中国药业,2009,18(17):22.
[8]杨 卿.高效液相色谱法测定复方缬沙坦片中缬沙坦的含量[J].山西医科大学学报,2009,40(12):1 093.
[9]Erk N.Simultaneous analysis of candesartan cilexetil and hydrochlorothiazide in human plasma and dosage forms using HPLC with a photodiode array detector[J].J Liq Chrom Relat Tech,2003,26(15):2 581.
猜你喜欢
世界最新医学信息文摘(2021年12期)2021-06-09 08:37:02
中华养生保健(2020年10期)2021-01-18 06:46:08
中华养生保健(2020年5期)2020-11-16 01:44:34
实用中西医结合临床(2015年7期)2015-02-28 16:30:39
湖北理工学院学报(2015年1期)2015-02-27 15:02:36
卫生职业教育(2014年16期)2014-05-16 03:48:28
精细石油化工(2014年3期)2014-03-14 03:01:12
中医研究(2014年2期)2014-03-11 20:28:18
河南医学研究(2014年1期)2014-02-27 14:51:16
