
盐酸溶液处理钒酸铋增强可见光催化活性及其机理
2012-03-06 04:43:36龙明策刘伊依陈渊源
物理化学学报 2012年12期
龙明策 万 磊 曾 曾 刘伊依 陈渊源
(上海交通大学环境科学与工程学院,上海200240)
1 引言
近年来含铋化合物由于具有较宽的响应光谱,在光催化领域受到广泛关注.黄色的钒酸铋(BiVO4)长期以来作为铋黄颜料被使用,由于单斜白钨矿的BiVO4具有光催化分解水和降解有机污染物的活性,1-3特别是其带隙宽度约为2.40 eV,对应吸收波长可以拓展到516 nm,相对于目前广泛研究的仅响应紫外光的纳米TiO2,具有明显的宽谱响应的优势,因此在光催化中具有很强的应用潜力.针对单一BiVO4粒径较大、活性较低的问题,研究者探索了包括超声法、4水热法、5-8溶剂热法9,10等在内的各种制备方法,以获得小粒径或优势晶面增长的高活性BiVO4纳米粒子;另一方面通过表面修饰或引入复合异质结构,如p/n异质结Co3O4/BiVO4、11,12贵金属修饰的Au/BiVO413,14等,均能获得显著提高的光催化活性.
另一方面氧卤化铋(BiOX,X=Cl、Br和I)作为一种新型光催化剂受到一定关注,相应的制备方法以及复合结构增强光催化活性的研究已有一些报道,15-17而相关氧化物在酸性条件下的相互转化规律已有初步研究.18-20如何通过光催化剂结构和组成的设计与调控获得更高能效的稳定光催化材料,仍是光催化研究所面临的挑战之一.21目前研究较多的方法主要集中在掺杂、表面贵金属修饰、优势晶面调控、复合异质结构等,对探索新的活性增强的改性途径具有重要理论价值和实际意义.本文发现盐酸水溶液处理BiVO4能显著提高其光催化活性,通过结构表征分析了处理前后BiVO4结构和组成的变化,进一步讨论了光催化活性增强的机理.
2 实验部分
2.1 催化剂的制备
硝酸铋、偏钒酸铵、HNO3、H2SO4、NH4Cl、NaCl、KCl、乙二醇均为分析纯,国药集团化学有限公司产品.
采用均匀沉淀法制备BiVO4:2,11将等摩尔的硝酸铋和偏钒酸铵分别溶解在200 mL浓度为1.84 mol·L-1的硝酸中,混合后得到二者浓度为0.2 mol· L-1.加入15 g尿素,在90°C持续搅拌8 h.反应后的黄色沉淀经过滤、水洗和干燥后,得黄色的BiVO4粉末备用.
酸溶液处理BiVO4的典型流程如下:将1 g的BiVO4粉末分散到50 mL浓度为0.1 mol·L-1的HCl水溶液中,在20°C反应6 h.悬浊液经过滤、水洗和干燥,得到最终的催化剂粉末.实验中研究了不同盐酸浓度和反应时间的影响,并与HNO3、H2SO4、NH4Cl和NaCl等处理的结果进行对照.
采用水热法制备BiOCl:16取5 mmol的硝酸铋和KCl溶解在45 mL的乙二醇中,混合后充分搅拌1 h,转入50 mL的水热反应釜在150°C保持16 h.生成的白色粉末经过滤、水洗和干燥后备用.
2.2 催化剂的表征
采用Rigaku D/Max2200/PC型X射线衍射仪研究样品的晶型和晶化度,具体条件为:Cu Kα线(λ= 0.154 nm),管电压40 kV,管电流20 mA,扫描范围10°-60°,扫描速率5(°)·min-1.样品的DRS谱在Lambda 950型紫外-可见分光光度计上测得,扫描范围200-800 nm,并由Kubelka-Munk函数转化为等价的吸收光谱F(R).采用Zeiss Ultra 55型扫描电镜对样品的粒径和形貌进行分析.采用XRF-1800顺序扫描型X射线荧光光谱仪定量分析Bi,V和Cl的含量.
2.3 光催化活性测试
以1000 W的氙灯作为光源,使用滤光片隔离波长小于400 nm的入射光,进行可见光催化活性的测试.通过光照下降解苯酚评价光催化活性:以100 mL烧杯作为反应器,将0.15 g催化剂样品分散于50 mL浓度为10 mg·L-1的苯酚水溶液中.光照前,样品先在避光条件下搅拌15 min以达到吸附平衡,取初始样;光照开始后,每隔30 min取反应液样品5 mL,用G5砂芯漏斗过滤,滤液通过4-氨基安替比林显色法测定苯酚浓度.
2.4 BiOCl的平带电位测试
采用悬浮液光电压法测定BiOCl的平带电位.22以全波长的350 W氙灯为光源,采用隔热滤光片滤除红外光.将80 mg的BiOCl粉末置于50 mL浓度为0.1 mol·L-1的KNO3溶液中,随后加入10 mg的甲基紫精(MV)Cl2.铂片电极作为工作电极,饱和甘汞电极作为参比电极.并持续通氮气,滴加HNO3调节pH值在3.0左右,待光电压数值稳定后,开始用经过N2吹脱的NaOH调节电解质溶液的pH值,并记录光电压随pH值的变化规律.
3 结果与讨论
3.1 光催化活性测试
采用浓度为0.2 mol·L-1的盐酸水溶液处理BiVO4,盐酸处理时间对产物光催化降解苯酚活性的影响如图1所示.从图1(A)中可以看出:对照实验显示在没有添加光催化剂时,苯酚在可见光照下不发生光解,浓度基本不发生变化;反应0 h的对照样,即均匀沉淀法制备的原始BiVO4,降解苯酚活性很弱,可见光催化反应2 h的苯酚降解率仅为14.2%,这是因为均匀沉淀法制备的BiVO4颗粒较大,光生载流子严重复合而导致活性较差;11经过盐酸溶液处理后,产物的光催化活性显著提高,处理6 h的催化剂在光催化降解苯酚2 h去除率达到41%,相对处理前活性提高了近3倍;而继续延长盐酸处理时间,活性没有显著增加,说明反应6 h后达到了平衡状态,催化剂不再发生变化.图1(B)是不同浓度的盐酸溶液处理BiVO4反应6 h后,催化剂降解苯酚的活性.由图可知,0.1 mol·L-1的盐酸处理后,催化剂表现出最佳光催化活性,光催化反应2 h的苯酚去除率达到49%.BiVO4粉末在盐酸水溶液中发生某种转化反应,反应随着盐酸浓度的增加而加速,但是继续提高盐酸浓度将导致BiVO4的溶出过快,显著破坏原有晶体结构,导致其光催化活性下降.过高浓度的盐酸(>0.5 mol·L-1)出现BiVO4显著溶解的现象.因此选定处理方法为采用0.1 mol·L-1的盐酸水溶液处理6 h,此时固体催化剂的质量没有明显变化,外观可见黄色表面略微发白,可见光催化降解苯酚的活性提高了3.5倍.
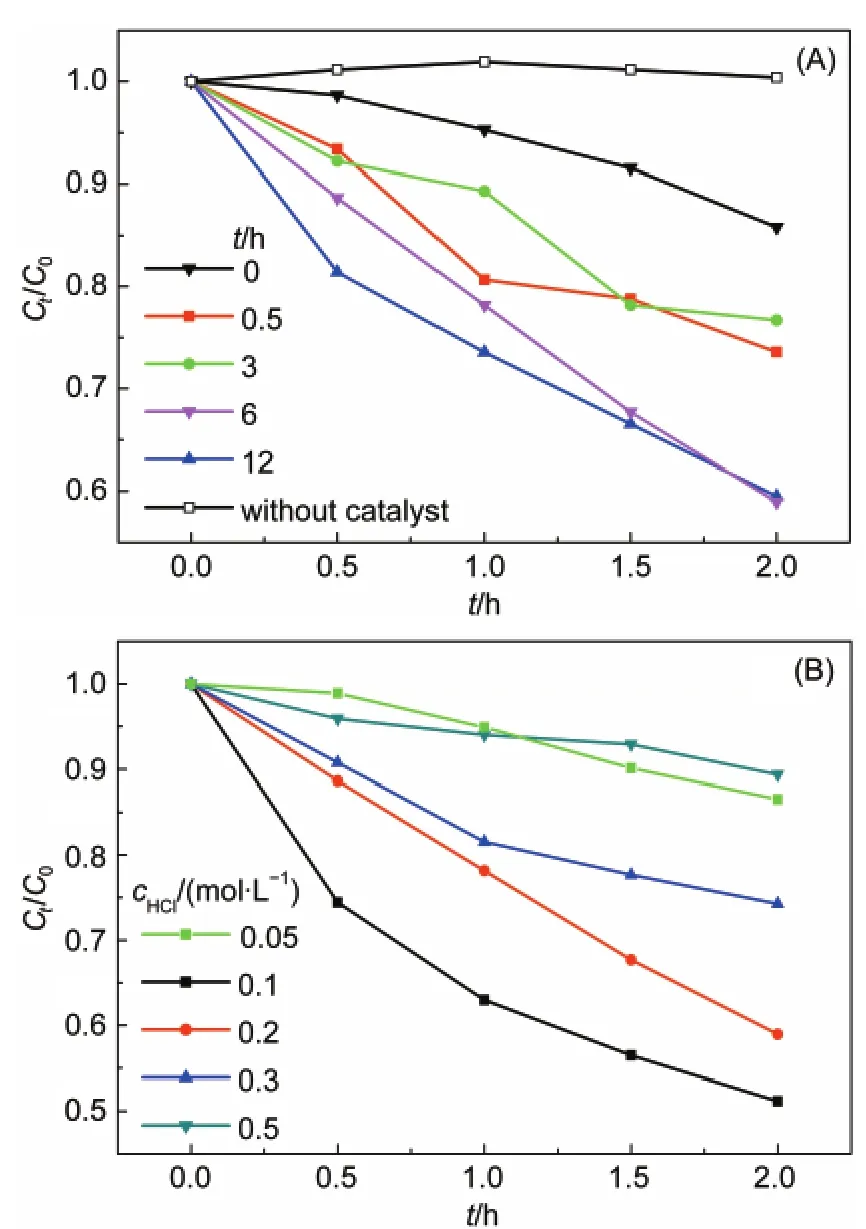
图1 不同盐酸处理条件对光催化降解苯酚活性的影响Fig.1 Influence of reaction conditions of BiVO4in HCl aqueous solution on the photocatalytic performance of phenol degradation(A)cHCl=0.2 mol·L-1;(B)t=6 h
为确定HCl处理引起BiVO4光催化活性增强的原因,比较了不同酸类型和氯化物的影响.采用相同H+浓度的盐酸、硝酸和硫酸,以及0.1 mol·L-1的NH4Cl和NaCl水溶液处理6 h作为对照实验,处理后的催化剂降解苯酚活性如图2所示.只有盐酸处理后表现出大大增强的光催化活性,而硝酸和硫酸处理,光催化活性甚至略微下降.这说明盐酸处理BiVO4增强光催化活性与溶液中的氯离子密切相关.同时可以看到采用氯化物NH4Cl和NaCl处理BiVO4,光催化活性也没有明显变化,说明H+和Cl-的协同作用使得BiVO4的结构或组成发生了关键变化,从而具有显著增强的光催化活性.将采用0.1 mol·L-1盐酸处理反应6 h的BiVO4样品记为T-B,进一步进行结构表征和机理分析.
3.2 结构表征
3.2.1 XRD分析
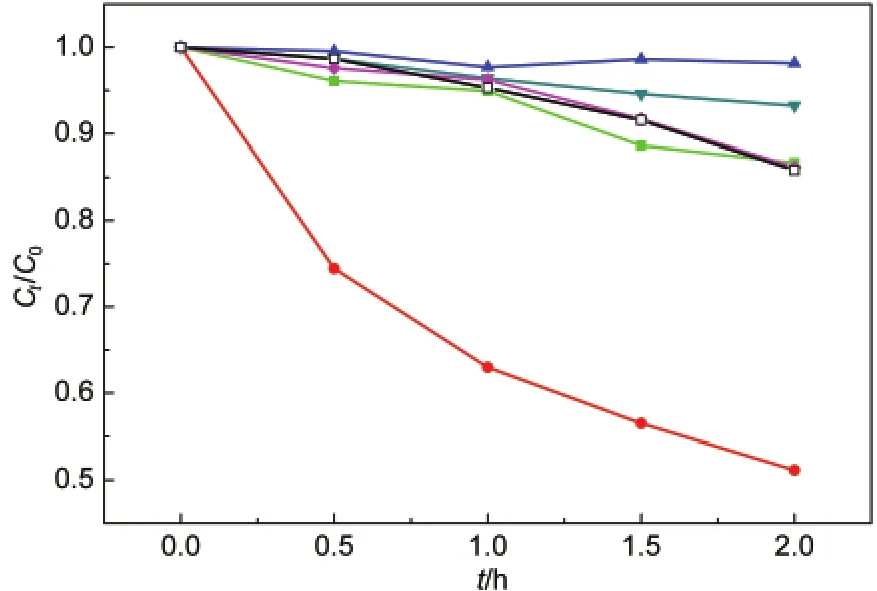
图2 BiVO4经不同酸和氯化合物处理后的光催化降解苯酚活性Fig.2 Photocatalytic performance of phenol degradation over the treated BiVO4in different acids or chlorides―□―BiVO4;―▼―0.1 mol·L-1HNO3;―▲―0.05 mol·L-1H2SO4;―●―0.1 mol·L-1HCl(T-B);―■―0.1 mol·L-1NH4Cl;―♦―0.1 mol·L-1NaCl;T-B:BiVO4sample treated in 0.1 mol·L-1HCl for 6 h

图3 催化剂样品的XRD谱图Fig.3 XRD patterns of various photocatalysts(A)BiVO4before and after treated in HCl aqueous solution;(B)BiOCl
图3是制备的BiVO4、BiOCl和BiVO4经盐酸处理后的T-B样品的XRD谱图.由图3可见,BiVO4的特征衍射峰(2θ=18.7°,28.6°,30.5°等)对应了标准卡片JCPDS 75-2480,23说明采用均匀沉淀法制备的BiVO4是单斜白钨矿结构.相对于BiVO4的其它两种晶相(四角白钨矿和四角锆石矿),单斜白钨矿具有更高的光催化活性.24采用水热法制备的BiOCl,其XRD图谱显示出特征衍射峰2θ值分别为25.9°、32.6°、33.5°,与BiOCl的标准卡片(JCPDS 73-2060)的结构一致,对应的晶面指数分别为(011)、(110)和(012).16而BiVO4经过盐酸处理后,T-B样品对应的单斜白钨矿BiVO4的所有特征峰未发生明显变化,同时在2θ=25.8°,33.4°处出现了两个较弱但是可分辨的特征衍射峰.比对BiOCl的特征图谱可知,这两个衍射峰分别对应了BiOCl的(011)和(012)晶面.16从峰强度可以看出,BiOCl的含量非常低.由此可以确定,在盐酸溶液的作用下,钒酸铋与溶液中的氯离子反应形成了少量氯氧化铋化合物.
3.2.2 紫外-可见漫反射吸收光谱分析
从外观上可以分辨催化剂粉末颜色略有差异,处理前的BiVO4为亮黄色;而处理后,黄色的粉末略带白色.图4是盐酸溶液处理前后BiVO4的紫外可见吸收光谱的比较.两条光学吸收曲线存在一定差异,首先处理后的样品光学吸收发生了蓝移,说明有宽带隙化合物掺杂其中干扰了BiVO4的光学吸收.同时可以看到波长在300-340 nm左右出现新的半导体吸收带,截止波长约在340 nm,接近实验制备的BiOCl的光学吸收截止波长347 nm(图4(A)).考虑到BiOCl和BiVO4均为间接带隙半导体,以(F(R)hv)0.5对hv做图,根据线性截距可以获得间接带隙宽度.其中主要成分的BiVO4的带隙宽度很容易获得,为2.34 eV.而BiOCl含量较少,以拐点处作水平线,与BiOCl吸收对应的线性部分延长线的交点所对应的横坐标作为带隙宽度,估算得到BiOCl的带隙宽度为3.66 eV,与文献报道的3.60 eV20接近.然而图4(B)显示BiOCl的带隙宽度约为3.43 eV,较低的带隙宽度数值可能与拖尾吸收有关,而拖尾吸收又可归因为催化剂颗粒表面的氧空位.
3.2.3 表面形貌分析

图4 紫外-可见漫反射吸收光谱和带隙宽度Fig.4 DRS UV-Vis spectra of samples and their estimated band gaps

图5 催化剂T-B的扫描电镜照片Fig.5 SEM images of catalyst T-B
图5是BiVO4经盐酸溶液处理的T-B样品的扫描电镜照片.通常均匀沉淀法制备的BiVO4颗粒表面平坦光滑,如图S1(见Supporting Information)所示,可以看出BiVO4颗粒聚集成粒径在微米尺度的无规则颗粒,但是表面基本光滑.13盐酸溶液处理后,颗粒仍呈现为无规则颗粒堆积的微米尺度形貌.在高倍放大下观察表面,可以看到颗粒表面形成了许多凹陷的沟壑,同时表面局部沉积了一些片状的异质化合物.这种片状结构与通常液相制备的BiOCl化合物的片状结构相似.16采用能量散射X射线(EDX)分析盐酸处理后催化剂的元素组成,结果见表1,可以看出元素氯以较低的浓度存在于处理后的催化剂中.这也说明经过盐酸水溶液处理后,样品表面形成了BiOCl化合物.
进一步采用X射线荧光光谱定量分析盐酸处理后样品T-B中BiOCl的含量,结果表明Bi和Cl元素的质量浓度分别为80.366%和0.286%,由此计算原子浓度比Bi:Cl为48,即BiOCl在T-B中的含量为2.08%.
3.3 光催化活性增强的机理探讨
根据上述分析可以看出,一定浓度的H+和Cl-协同作用改变了BiVO4的结构与组成.BiVO4可以溶解在强酸中,因此可推测一定浓度的H+存在条件下,将对BiVO4表面产生刻蚀破坏作用,使之以BiO+和VO3-的形式溶出,同时存在二者重新结晶的动态平衡.另一方面由热力学数据可知,BiO+在水溶液中的吉布斯自由能为-147 kJ·mol-1,而固态的BiOCl为-322 kJ·mol-1.25因此当Cl-存在时,BiO+有自发转化为BiOCl的趋势,从而使得BiVO4逐渐刻蚀溶出并转化为BiOCl.因此通过控制盐酸浓度和反应时间,可以控制BiVO4的刻蚀和BiOCl的生成速度与程度,从而经过溶出与沉积过程,最终形成了T-B样品的结构和组成,即具有凹陷沟壑表面的BiVO4与片状BiOCl的复合物.
BiVO4经盐酸溶液处理后,增强的光催化活性可能源于两个方面:(1)BiVO4表面形成的凹陷沟壑,因为粗糙化的表面有利于光生电荷分离,使得光催化活性优于平坦光滑表面;(2)BiVO4与BiOCl形成异质结,促进光生电荷分离,提高光催化活性.为进一步确定光催化活性增强的原因,对BiVO4与BiOCl的异质结构进行了探讨.
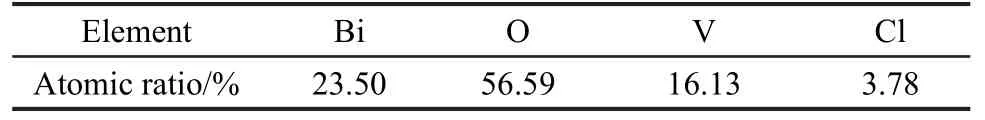
表1 EDX测试显示的催化剂T-B中各元素含量Table 1 Ratios of elements over the catalyst T-B by energy dispersive X-ray measurement
不同半导体化合物构成复合结构,必须在能带位置合适的前提下才能促进光生电荷的迁移与分离.为此采用悬浮液光电压法测定了BiOCl的平带电位,并与BiVO4进行比较.悬浮液光电压法是测试半导体化合物平带电位最常用的方法之一,该方法采用氧化还原电位不随pH值变化的电子受体为指示剂,记录光电压随pH值的变化,通过获取光电压突变点pH0并采用公式(1)计算任意pH值下的半导体化合物的平带电位(Efb):22

图6 BiOCl的光电压随悬浮液pH值的变化Fig.6 Photovoltage vs suspension pH value measured for BiOCl
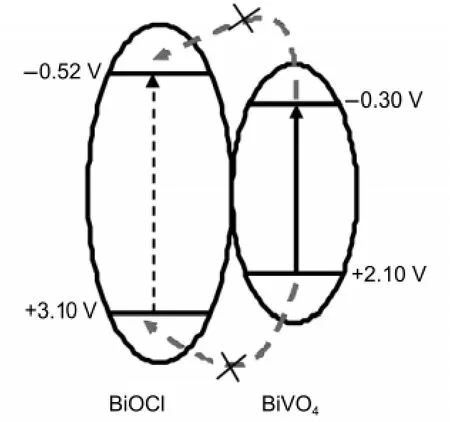
图7 BiOCl和BiVO4能级位置及光生电荷迁移原理图Fig.7 Energy diagram of BiOCl and BiVO4and photogenerated charges transfer process
式中E0是单电子受体的标准氧化还原电位,本实验采用甲基紫精(MV2+,E0=-0.45 V)作为单电子受体; k是电位随pH值变化的常数,其值通常为0.059 V/ pH.图6是BiOCl平带电位的测试曲线,采用Sigmoidal函数拟合获得pH0为5.73,根据公式(1)可以计算在pH=7时,BiOCl的平带电位为-0.52 V.若应用BiOCl的带隙宽度为3.60 eV估算,则价带位置在3.10 V,价带位置较正,说明BiOCl形成的空穴将具有较强的氧化能力.采用悬浮液光电压法测定的BiVO4在pH=7时的平带电位为-0.30 V,价带位置在2.10 V.12故此可做出图7所示的能级结构示意图,可以看出BiOCl的导带位置比BiVO4更负,而价带位置比BiVO4更正,因此在可见光照下,BiVO4激发产生的电子和空穴均不能转移到BiOCl,也就是说经过盐酸处理后,BiOCl的生成以及形成的异质结构不能发挥促进光生电荷分离的作用.
如果两种半导体光催化剂的能级位置适合光生载流子在异质组分上的分离,则两种混合颗粒在光催化反应中将发生颗粒间的电子转移现象,被称为颗粒间电子转移(IPET)效应.26为进一步否定异质结构促进光生电荷分离的推测,以一定比例(质量比)混合的BiVO4和BiOCl粉末为催化剂,测定混合颗粒的可见光光催化活性,如图8所示.尽管BiOCl的带隙较宽,但是在λ>400 nm的可见光条件下仍存在一定光催化活性,3 g·L-1的催化剂光照2 h苯酚降解率约为27%,这可能与BiOCl表面氧空位引起的响应性能有关.然而混合的BiVO4与BiOCl没有表现出增强的光催化活性,说明二者颗粒之间不存在IPET效应,这与前面能带位置的分析是一致的.
Kudo等27,28研究发现,采用镧或者碱土金属掺杂处理后,NaTaO3颗粒表面出现梯状结构,这种表面粗糙化的颗粒光催化活性优于平坦表面,因为光生电荷在凹凸不平表面更容易分离,使得光催化氧化反应发生在凹位,而光催化还原反应发生在凸位.BiVO4经过盐酸溶液处理后,由于刻蚀溶出和表面沉积,使得在光滑平坦的BiVO4表面形成了大量的凹陷沟壑,这种粗糙化结构使得BiVO4表面产生的光生电荷更易于分离,从而表现出增强的光催化活性.类似表面处理方法可能成为增强大颗粒光催化材料的光催化活性的一种有效而简易的途径.
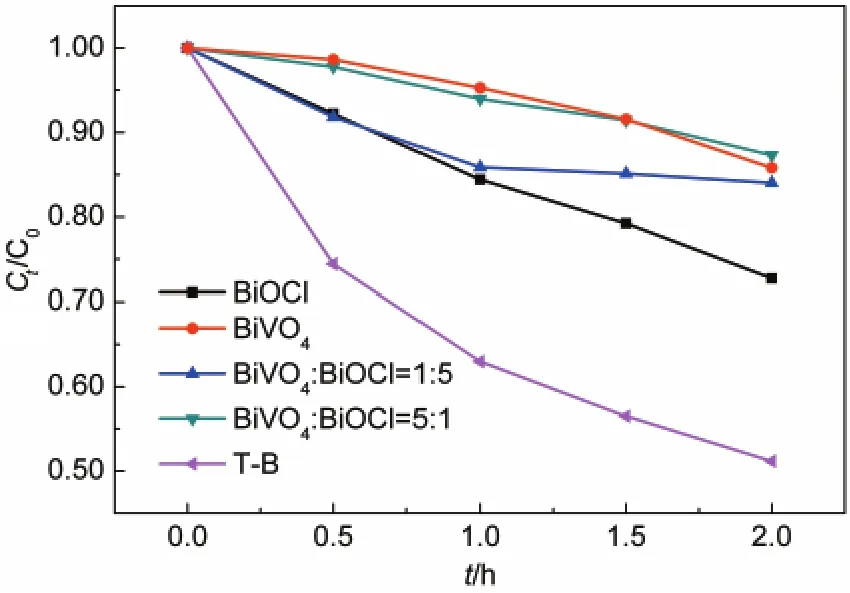
图8 不同质量比的BiVO4与BiOCl颗粒混合物及T-B降解苯酚活性Fig.8 Photocatalytic performance of phenol degradation over BiVO4and BiOCl mixed particles(mass ratio)and sample T-B
4 结论
发现盐酸水溶液处理BiVO4可以增强其光催化活性.在0.1 mol·L-1盐酸溶液中处理6 h,BiVO4的可见光催化降解苯酚的活性提高了3.5倍.结构表征结果显示,经处理后少量BiVO4转化为BiOCl,形成了具有凹陷沟壑表面的BiVO4与片状BiOCl的复合异质结构,其中BiOCl含量为2.08%.H+和Cl-的协同作用,使得BiVO4表面部分溶出并以BiOCl沉积,从而获得活性增强的光催化剂.采用悬浮液光电压法测定BiOCl平带电位为-0.52 V(pH=7).能带位置分析和混合颗粒光催化性能测试表明,BiVO4与BiOCl二者间不存在颗粒间电子转移效应.盐酸处理后增强的光催化活性主要源于BiVO4表面形成了有助于光生电荷迁移的凹凸不平结构.
Supporting Information: available free of charge via the internet at http://www.whxb.pku.edu.cn.
(1) Kudo,A.;Ueda,K.;Kato,H.;Mikami,I.Catal.Lett.1998,53, 229.doi:10.1023/A:1019034728816
(2) Kohtani,S.;Makino,S.;Kudo,A.;Tokumura,K.;Ishigaki,Y.; Matsunaga,T.;Nikaido,O.;Hayakawa,K.;Nakagaki,R.Chem. Lett.2002,31,660.
(3)Kohtani,S.;Tomohiro,M.;Tokumura,K.;Nakagaki,R.Appl. Catal.B:Environ.2005,58,265.doi:10.1016/j.apcatb. 2004.12.007
(4)Zhou,L.;Wang,W.Z.;Liu,S.W.;Zhang,L.S.;Xu,H.L.;Zhu, W.J.Mol.Catal.A:Chem.2006,252,120.doi:10.1016/j. molcata.2006.01.052
(5) Zhang,L.;Chen,D.R.;Jiao,X.L.J.Phys.Chem.B 2006,110, 2668.doi:10.1021/jp056367d
(6)Wang,D.;Jiang,H.;Zong,X.;Xu,Q.;Ma,Y.;Li,G.;Li,C. Chem.Eur.J.2011,17,1275.doi:10.1002/chem.v17.4
(7)Xi,G.;Ye,J.Chem.Commun.2010,46,1893.
(8) Cheng,B.;Wang,W.G.;Shi,L.;Zhang,J.;Ran,J.R.;Yu,H.G. Int.J.Photoenergy 2012,797968.
(9) Ren,L.;Jin,L.;Wang,J.B.;Yang,F.;Qiu,M.Q.;Yu,Y. Nanotechnology 2009,20,115603.doi:10.1088/0957-4484/20/ 11/115603
(10)Jiang,H.Y.;Dai,H.X.;Meng,X.;Zhang,L.;Deng,J.G.;Ji,K. M.Chin.J.Catal.2011,32,939. [蒋海燕,戴洪兴,孟 雪,张 磊,邓积光,吉科猛.催化学报,2011,32,939.]doi: 10.1016/S1872-2067(10)60215-X
(11)Long,M.C.;Cai,W.M.;Cai,J.;Zhou,B.X.;Chai,X.Y.;Wu, Y.H.J.Phys.Chem.B 2006,110,20211.doi:10.1021/ jp063441z
(12)Long,M.C.;Cai,W.M.;Kisch,H.J.Phys.Chem.C 2008,112, 548.doi:10.1021/jp075605x
(13) Long,M.C.;Jiang,J.J.;Li,Y.;Cao,R.Q.;Zhang,L.Y.;Cai, W.M.Nano-Micro Lett.2011,3,171.
(14) Cao,S.W.;Yin,Z.;Barber,J.;Boey,F.Y.;Loo,S.C.;Xue,C. ACS Appl.Mater.Interfaces 2012,4,418.doi:10.1021/ am201481b
(15)Huang,W.L.;Zhu,Q.S.J.Comput.Chem.2008,30,183.
(16) Zhang,X.;Ai,Z.H.;Jia,F.L.;Zhang,L.Z.J.Phys.Chem.C 2008,112,747.doi:10.1021/jp077471t
(17)Yu,C.L.;Cao,F.F.;Shu,Q.;Bao,Y.L.;Xie,Z.P.;Yu,J.C.; Yang,K.Acta Phys.-Chim.Sin.2012,28,647.[余长林,操芳芳,舒 庆,包玉龙,谢志鹏,Yu,Y.J.,杨 凯.物理化学学报, 2012,28,647.]doi:10.3866//PKU.WHXB201201051
(18)Wang,W.;Huang,F.;Lin,X.Scripta Mater.2007,56,669.doi: 10.1016/j.scriptamat.2006.12.023
(19) Chai,S.Y.;Kim,Y.J.;Jung,M.H.;Chakraborty,A.K.;Jung, D.;Lee,W.I.J.Catal.2009,262,144.doi:10.1016/j. jcat.2008.12.020
(20) Chang,X.;Yu,G.;Huang,J.;Li,Z.;Zhu,S.;Yu,P.;Cheng,C.; Deng,S.;Ji,G.Catal.Today 2010,153,193.doi:10.1016/j. cattod.2010.02.069
(21) Kubacka,A.;Fernandez-Garcia,M.;Colon,G.Chem.Rev. 2012,112,1555.doi:10.1021/cr100454n
(22) Roy,A.M.;De,G.C.;Sasmal,N.;Bhattacharyya,S.S.Int.J. Hydrog.Energy 1995,20,627.doi:10.1016/0360-3199(94) 00105-9
(23) Li,B.X.;Wang,Y.F.;Liu,T.X.Acta Phys.-Chim.Sin.2011, 27,2946.[李本侠,王艳芬,刘同宣.物理化学学报,2011,27, 2946.]doi:10.3866/PKU.WHXB20112946
(24)Kudo,A.;Omori,K.;Kato,H.J.Am.Chem.Soc.1999,121, 11459.doi:10.1021/ja992541y
(25) Dean,J.A.Langeʹs Chemistry Handbook,13rd ed.;Science Press:Beijing,1991;p 9-9;translated by Sang,J.F.,Cao,S.J., Xing,W.M.,Zheng,F.Y.,Lu,X.M.[Dean,J.A.兰氏化学手册.尚久方,操时杰,辛无名,郑飞勇,陆晓明,林长青译.北京:科学出版社,1991:9-9.]
(26) Robert,D.Catal.Today 2007,122,20.doi:10.1016/j. cattod.2007.01.060
(27)Kato,H.;Asakura,K.;Kudo,A.J.Am.Chem.Soc.2003,125, 3082.doi:10.1021/ja027751g
(28) Iwase,A.;Kato,H.;Okutomi,H.;Kudo,A.Chem.Lett.2004, 33,1260.doi:10.1246/cl.2004.1260
猜你喜欢
——潘桂棠光生的地质情怀
沉积与特提斯地质(2021年2期)2021-07-20 06:33:26
云南化工(2020年11期)2021-01-14 00:50:54
物理化学学报(2017年3期)2017-03-11 00:25:30
昭通学院学报(2016年5期)2016-02-24 10:51:12
合成化学(2015年4期)2016-01-17 09:01:27
淮南师范学院学报(2015年3期)2015-03-22 01:16:19
河北科技大学学报(2015年5期)2015-03-11 16:16:34
石家庄铁道大学学报(自然科学版)(2015年3期)2015-02-28 15:05:43
无机化学学报(2014年4期)2014-02-28 17:31:23
应用技术学报(2014年1期)2014-02-28 14:52:11
