
凡德他尼的合成工艺研究
2012-01-15 03:51赵玲,杨博
武汉轻工大学学报 2012年2期
赵 玲,杨 博
(1.武汉工业学院生物与制药工程学院,湖北武汉430023;2.南京安格医药化工有限公司,江苏南京210009)
近年来,分子肿瘤学、分子药理学的发展使发生肿瘤的本质正在逐步被阐明,大规模快速筛选、组合化学、基因工程等先进技术的发明和应用加速了药物开发进程,抗肿瘤药物的研发已进入崭新的时代。细胞内信号转导是目前国内、外的研究热点之一,原癌基因与抗癌基因产物的分布涉及从细胞膜外至细胞膜内的全部信号传递系统,膜外信号及膜的信号传递是信号转导过程中首要的关键步骤,最典型的例子是生长因子与其受体结合而激活受体。酪氨酸激酶是最常见的生长因子受体,通过阻断酪氨酸激酶可破坏肿瘤细胞的信号传递,从而达到抗肿瘤的目的。受体酪氨酸激酶抑制剂包括单靶点酪氨酸激酶抑制剂和多靶点酪氨酸激酶抑制剂。细胞信号传导过程复杂,涉及多蛋白结构和功能变化的生化系统,由单一因素的过表达来判断是否有肿瘤生长也许并不全面,试图通过单靶点药物阻断某个受体来阻断肿瘤细胞所有信号转导,疗效可能不够客观,也有可能引起耐药,于是产生了多靶点药物的概念。目前有关多靶点受体酪氨酸激酶抑制剂的研究非常活跃,出现了一些新药,有的已取得了重大突破性进展,为肿瘤治疗开启了希望之门。目前已上市应用于临床的有索拉非尼(sorafenib)、尼 罗 替 尼( nilotinib)、舒 尼 替 尼(sunitinib)、凡德他尼(vandetanib)、达沙替尼(dasatinib)、拉帕替尼(lapatinib)、阿西替尼(axitinib) 和帕唑帕尼(pazopanib)[1]。
凡德他尼(Vandetanib 1),化学名为4-(4-溴-2-氟苯胺基)-6-甲氧基-7-[(1-甲基哌啶-4-基)甲氧基]喹唑啉,商品名为Caprelsa,是由英国阿斯利康公司研发的的口服小分子多靶点酪氨酸激酶抑制剂,对血管上皮生长因子受体(VEGFR)和表皮生长因子受体(EGFR)均有抑制作用,不仅作用于肿瘤细胞EGFR、VEGFR和RET酪氨酸激酶,还可选择性地抑制其他的酪氨酸激酶以及丝氨酸/苏氨酸激酶。2011年4月在美国上市,2012年2月在欧洲上市,用于治疗不能切除、局部晚期或转移的有症状或进展的髓样甲状腺癌[2]。
1 合成路线分析
文献报道了凡德他尼及其衍生物的合成[3-8],主要有两条路线:路线 1,以香草酸为原料,经烷基化、硝化、还原、环合得到7-苄氧基-6-甲氧基-3,4-二氢喹唑啉-4-酮,再经氯化、缩合、脱保护、缩合反应得到7-[(1-叔丁氧羰基)哌啶-4-甲氧基]-4-(4-溴-2-氟苯胺)-6-甲氧基喹唑啉,最后在甲醛甲酸条件下发生脱保护、还原胺化反应得到1(图1)。路线2,以1-叔丁氧碳基-4-甲磺酰基氧甲基哌啶为起始原料,直接与香草酸甲酯发生取代反应,然后再经脱保护,还原氨化、还原、环合,氯化,氨化得到1(图2)。
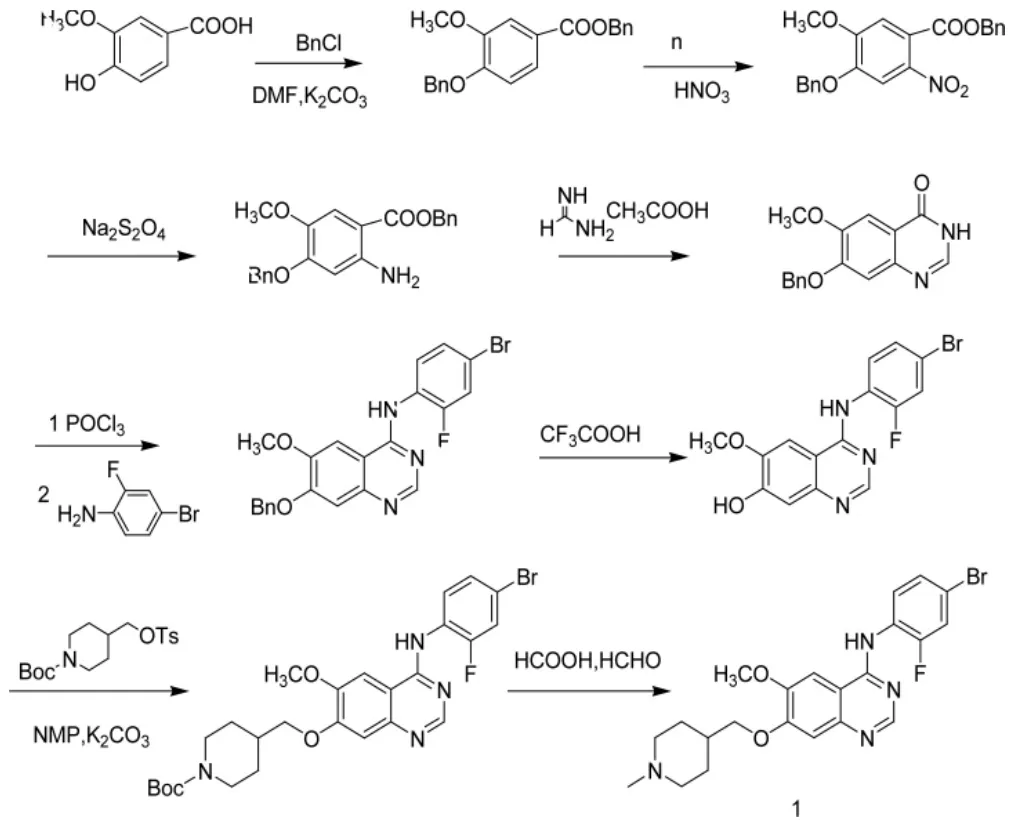
图1 路线1
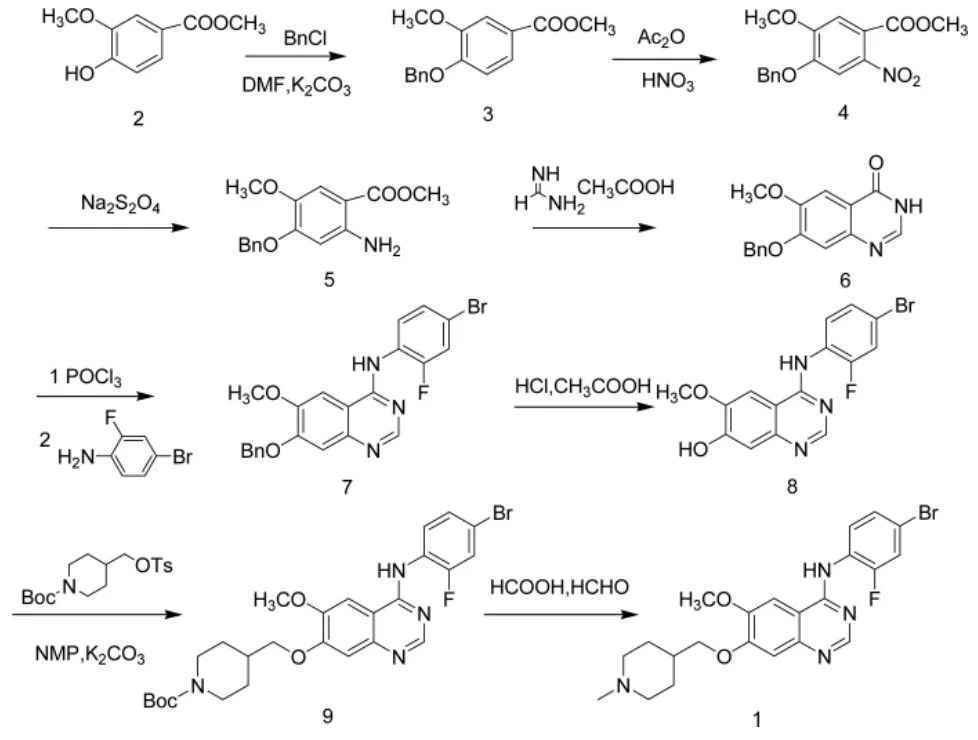
图2 路线2
本文通过文献调研和预实验,确定采用以商业化供应的香草酸甲酯起始原料的合成工艺路线(图3)。化合物4的合成,文献[3]用硝酸/醋酸体系进行硝化,硝化温度高,反应慢,且不完全,本研究改用乙酸酐/硝酸条件进行硝化,后处理将反应液直接倒入冰水中即可析出产品,反应条件温和,收率高。化合物5的合成,文献[3-4]用乙腈做溶剂,本研究改用乙醇,降低了溶剂成本。制备6时,本研究用甲醇做溶剂替换文献[3]中异丁醇,并采用分批加入的方式加入醋酸甲脒,降低了溶剂成本的同时,减少了醋酸甲脒的用量。制备8时,文献[3-4]用三氟乙酸脱苄基,污染大,本研究改用浓盐酸/冰醋酸脱苄基,成本降低,且对设备腐蚀性小。制备9时,发现需要在适当温度下搅拌一段时间,否则氮取代副产物将会增多。制备1时,文献[3-4]用到甲酸,并没有提到浓度,通过实验摸索,我们采用价格低廉的80%的甲酸作为反应试剂,同时对1的纯化工艺进行了摸索,发现乙醇/水(4∶1)重结晶效果最好。改进后的工艺原料价廉易得,条件温和,后处理简便,成本低,适合工业化生产,以2计总收率约为43%。
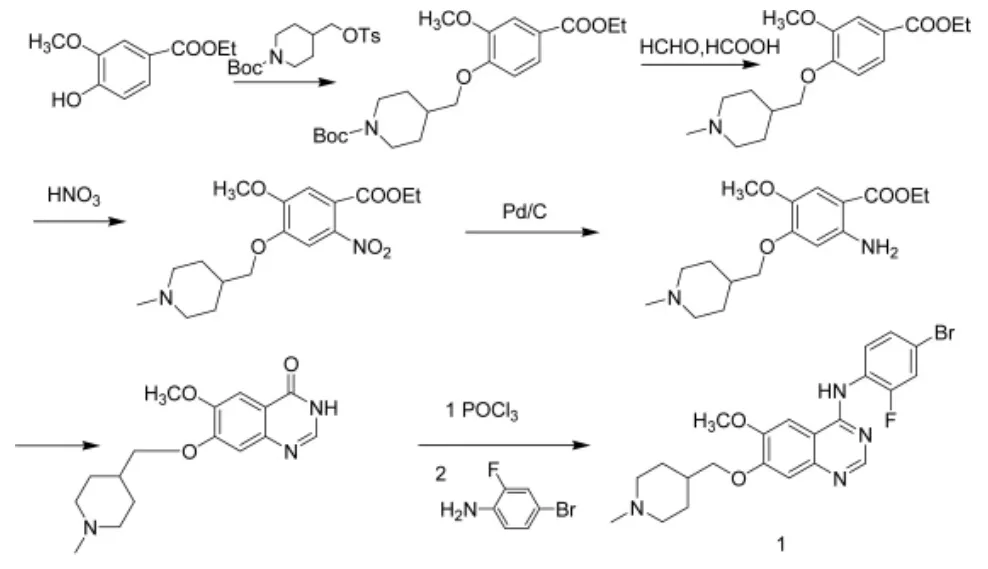
图3 本论文采用的合成路线
2 实验部分
2.1 仪器和试剂
熔点仪为MP-500显微熔点仪(温度计未经校正);核磁共振仪为Brucker AV-300,AV-500型核磁共振波谱仪;质谱仪为Agilent 1100-MSD-Trap型质谱仪;高效液相色谱仪为岛津10A;所用化学试剂均为国产分析纯试剂,流动相为色谱纯试剂。
2.2 3-甲氧基-4-苄氧基香草酸甲酯(3)的合成
2(546 g,3 mol)和碳酸钾 (414 g,3 mol)溶于DMF(1500 mL)中,加热至100℃反应10 h,冷却过滤,回收大部分溶剂,加水过滤,干燥得白色固体(793 g,97%)。mp 81—82℃ (文献[9]:80.5—82℃)。1H NMR(DMSO-d6)δ:3.83(s,6H),5.18(s,2H),7.13(d,1H),7.27-7.49(m,6H),7.53(dd,1H)。
2.3 2-硝基-4-苄氧基-5-甲氧基苯甲酸甲酯(4)的合成
3(160 g,0.59 mol)加入乙酸酐 (200 mL)中,冰浴下滴加浓硝酸 (40 mL),滴毕,30℃反应3 h,反应液倒入冰水 (1000 mL)中,过滤,水 (500 mL)洗涤,干燥得淡黄色固体 (65 g,93%),mp 125—126 ℃(文献[9]:126—127 ℃)。1H NMR(DMSO-d6) δ:3.82(s,3H),3.92(s,3H),5.23(s,2H),7.33-7.54(m,6H),7.77(s,1H)。
2.4 2-氨基-4-苄氧基-5-甲氧基苯甲酸甲酯(5)的合成
将4(60 g,189 mmol)溶于乙醇 (300 mL)中,加入保险粉(150 g)的水(200 mL)溶液,滴加50%的氢氧化钠溶液(50 mL),保持PH至6,加毕,加热回流2h,冷却,加入浓盐酸 (20 mL),加热回流1 h后,冰浴下滴加50%氢氧化钠溶液 (50 mL)调节PH至9,过滤,40℃鼓风干燥得黄色固体 (46 g,85%),mp 130—131 ℃(文献[10]:132—133 ℃)。
2.5 7-苄氧基-6-甲氧基-3,4-二氢喹唑啉-4-酮(6)的合成
将5(120 g)、醋酸甲脒 (80 g)和甲醇 (600 ml)加入反应瓶中,加热回流6 h,冷却过滤,得类白色固体6(107 g,90%),mp 266—267 ℃(文献[11]:266 ℃)。1H NMR(DMSO-d6)δ:12.01(brs,1H),7.98(s,1H),7.33-7.50(m,6H),7.21(s,1H),5.26(s,2H),3.89(s,3H)。
2.6 7-苄氧基-4-(4-溴-2-氟苯胺)-6-甲氧基喹唑啉(7)的合成
将6(42 g,0.15 mol)、甲苯 (500 mL)、三乙胺(18 g,0.18 mol)和三氯氧磷 (28 g,0.18 mol)加入到反应瓶中,升温至90℃反应2 h,冷却至60℃,加入2-氟-4-溴苯胺 (34 g,0.18 mol),升温至80℃反应2 h,冷却至室温,过滤,干燥得黄色固体(64.5 g,95%),mp 247—248 ℃。1H NMR(DMSO-d6)δ:9.47(s,1H),8.35(s,1H),,7.86(s,1H),7.50-7.34(m,8H),7.26(s,1H),5.27(s,2H),3.94(s,3H)。
2.7 7-羟基-4-(4-溴-2-氟苯胺)-6-甲氧基喹唑啉(8)的合成
将7(50 g,0.11 mol)、冰醋酸 (300 ml)和盐酸(100 ml)加入反应瓶中,加热回流3 h,冷却,加水(300 ml),过滤,40℃真空干燥,得类白色固体 (36 g,90%),mp 213—214℃。1H NMR(DMSO-d6)δ:8.7(s,1H),8.3(s,1H),7.7(m,1H),7.5(m,2H),7.4(s,1H),4.0(s,3H)。
2.8 7-[(1-叔丁氧羰基)哌啶-4-甲氧基]-4-(4-溴-2-氟苯胺)-6-甲氧基喹唑啉(9)的合成
将8(25 g,69 mmol)碳酸钾 (17g,0.12 mol),NMP(200 ml),加入到反应瓶中,50℃搅拌1 h,加入1-叔丁氧碳基-4-甲磺酰基氧甲基哌啶(26g,70mmol)升温至90℃反应3 h,冷却,加水(200 ml),过滤,干燥得黄色固体(34 g,88%),mp213—214 ℃。1H NMR(DMSO-d6)δ:9.51(brs,1H),8.33(s,1H),7.8(d,1H),7.65(d,1H),7.41-7.50(m,2H),7.03(s,1H),4.01(m,4H),3.95(s,3H),2.71-2.93(m,2H),2.01-2.12(m,1H),1.81-1.84(d,2H),1.44(s,9H),1.11-1.33(m,2H)。
2.9 凡德他尼(1)的合成
将 9(20 g,36 mmol),80% 甲酸 (27ml),37%甲醛(5.64 g,70 mmol)加入反应瓶中,升温至80℃反应6 h,冷却,加入50%氢氧化钠溶液(60 ml),调PH至12,冷却过滤,干燥,甲醇/水(4∶1)重结晶,得白色固体 (14 g,83%),纯度99.7%,[HPLC归一化法:色谱柱 C18(4.6 mm ×250 mm,5 μm),流动相甲醇-0.05乙酸铵(70∶30,三乙胺调至pH 8.0),检测波长 247 nm,流速 1.0 mL/min,柱温25℃],mp 240—242 ℃(文献[7]:126—127 ℃)。1H NMR(DMSO-d6)δ:9.53(s,1H),8.34(s,1H),7.79(s,1H),7.63-7.67(d,1H),7.56-7.44(m,2H),7.17(s,1H),3.99-4.04(d,2H),3.94(s,3H),2.77-2.80(m,2H),2.16(s,3H),1.74-1.9(m,5H),1.29-1.41(m,2H),LC-ESI-MS(m/z):475[M+H]+。
3 结论
国内外多靶点受体酪氨酸激酶抑制剂的研发方兴未艾,已上市的国内外多靶点受体酪氨酸激酶抑制剂在中国目前都有化合物专利保护,前瞻性地开展其合成工艺研究,有利于为该领域的仿制和创制工作做好技术储备和经验积累。本研究对新型多靶点受体酪氨酸激酶抑制剂凡德他尼的合成工艺文献报道进行了甄选和改进,在硝化反应中采用醋酐硝酸条件,克服了醋酸硝酸反应温度高,反应不完全的缺点;在脱苄基过程中,采用盐酸替换污染较大的三氟乙酸,并对各步反应和后处理做了适当优化。所研制的合成工艺路线各步收率均较高,而且操作简便,无需特殊试剂和条件,绿色环保,预期适合工业化生产的要求。
[1] 唐海涛,陈国广,方正,等.多靶点受体酪氨酸激酶抑制剂的研究进展[J].中国新药杂志,2009,18(6):502-505.
[2] 张兰平.口服抗癌药 Vandetanib[J].药学进展,2007,31(4):190-191.
[3] Jörgen B,David G M,John H P,et al.Chemical process:WO,200736713[P].2007-04-05.
[4] Thomas G G,Sepehr S,Manoucherhr S.Substituted quinazoline inhibitors of growth factor receptor tyrosine kinases:WO,2010028254[P].2010-03-11.
[5] Hennequin L F,Stokes E,Thomas A P,et al.Novel 4-Anilinoquinazolines with C-7 basic side chains:design and structure activity relationship of a series of potent,orally active,VEGF receptor tyrosine kinase inhibitors[J].Journal of Medicinal Chemistry,2002,45(6):1300-1312.
[6] Laurent Francois Andre H,Elaine Sophie Elizabeth S,Andrew Peter T.Preparation of 4-anilino-7-piperidinyloxy quinazolines as vascular endothelial growth factor inhibitors:WO,2001032651[P].2001-05-10.
[7] 刘宇,夏超,刘媛.抗癌药凡德他尼的合成[J].中国抗生素杂志,2011,36(12):917-920.
[8] Tasler S,Müller O,Wieber T,et al,Substituted 2-arylbenzothiazoles as kinase inhibitors:Hit-to-lead optimization[J].Bioorganic& Medicinal Chemistry,2009,17(18):6728-6737.
[9] Wang T,Lui A S,Cloudsdale L S.A novel route to pyrrolo[2,1-c][1,4]benzodiazepin-5-ones.formal total synthesis of( ±)-DC-81[J].Org Lett,1999,1(11):1835-1837.
[10] Kamal A,Markandeya N,Shankaraiah N,et al,Chemoselective aromatic azido reduction with concomitant aliphatic azide employing Al/Gd triflates/naI and ESI-MS mechanistic studies[J].Chemistry--A European Journal 2009,15(29):7215-7224.
[11] Hennequin L F,Thomas A P,Johnstone C,et al.Design and structure-activity relationship of a new class of potent VEGF receptor tyrosine kinase inhibitors[J].J Med Chem,1999,42(26),5369-5389.
猜你喜欢
能源化工(2022年1期)2023-01-14
能源化工(2021年6期)2021-12-30
纺织检测与标准(2021年3期)2021-12-03
铜仁学院学报(2018年6期)2018-07-05
中成药(2017年4期)2017-05-17
中成药(2017年3期)2017-05-17
中国洗涤用品工业(2016年2期)2016-02-28
中国塑料(2015年2期)2015-10-14
云南中医学院学报(2015年2期)2015-07-31
现代检验医学杂志(2014年3期)2014-02-02
