
适用于HCCI燃烧研究的甲苯参比燃料化学动力学简化模型
2011-12-12 02:43张庆峰郑朝蕾何祖威
物理化学学报 2011年3期
张庆峰 郑朝蕾 何祖威 王 迎
(重庆大学低品位能源利用技术及系统教育部重点实验室,重庆400030)
1 Introduction
Homogeneous charge compression ignition(HCCI)combustion process holds the promise of both good fuel economy and very low emissions of nitrogen oxides and soot.Therefore, HCCI is considered as a high-efficiency alternative to sparkignited(SI)gasoline operation and as a low-emission alternative to traditional diesel compression ignition(CI)combustion. In a HCCI engine,a homogeneous mixture of fuel and air is compressed sufficiently to raise the mixture temperature above its ignition point.Ignition is distributed throughout the cylinder volume,starting at the hottest regions.As the mixture burns, the expanding gases cause further compression of the unburned mixture and subsequent ignition without flame propagation.More detailed information about progress and recent trends in HCCI engines,as dicussed by Yao et al.1
The combustion in HCCI engine is controlled by the chemical kinetics.2Hence,the chemical kinetics modeling is one of an important tool to the research of HCCI combustion.The combustion process is initiated by autoignition in HCCI engines.Autoignition depends on the pressure and temperature history of the fuel-air mixture.For the characterization of fuels the autoignition delay is a significant observable and is a critical parameter for HCCI engines.Tanaka et al.3performed experiments on HCCI combustion in a rapid compression machine with complex fuels,such as cyclic paraffins,olefins,and aromatics,which exist in petroleum-based fuels.They concluded that ignition delay time and burn rate could be independently controlled using various fuel mixtures and additives for HCCI combustion.
Gasoline is one of the most important fuels in internal combustion engine,and it is a complex mixture of hundreds of hydrocarbons,such as paraffins,olefins,aromatics,and cycloalkanes.Unfortunately,it is not currently possible to represent the complex chemistry of full blend of gasoline in a detailed chemical kinetics model.In general,the term surrogate denotes a simpler representation of a fully-blended fuel.The use of iso-octane is the simplest gaoline surrogate.Binary blends of iso-octane and n-heptane,pimary reference fuel(PRF)is applied widespread as gasoline surrogates for variable octane number fuel.With rapid growth of the HCCI combustion research,the study on chemical kinetics mechanism of iso-octane and PRF become very active.However,it is clear that the values of octane number are insufficient in characterzing autoignition behavior under HCCI combustion conditions.Due to differing physical and chemical properties,the behavior of PRF mixtures in homogenous charge and spark ignition engines is significantly different than that of gasoline itself.Consequently,more complex mixtures of individual species representing different molecular groups have been suggested as surrogate fuels for gasoline.As explained above,gasoline contains significant quantities of aromatic hydrocarbons.Toluene is the largest single aromatic component of the majority of fuels and the mole fraction is as high as 35%in premium fuels, and toluene is formed during the oxidation of other hydrocarbons and commercial fuels.4Therefore,toluene should be concerned.The relative importance of fuel components for gasoline,diesel and jet fuel surrogates were reported by Pitz et al.,5the result shows that a ternary mixture of iso-octane,n-heptane and toluene is suitable as a gasoline surrogate fuel for the HCCI engine simulation.This ternary blend is usually called toluene reference fuel(TRF).
The prior published mechanisms for iso-octane and n-heptane developed by Curran et al.6,7were minimized,optimized and combined to reproduce PRF results.Chaos et al.8constructed a detailed chemical kinetic mechanism for the TRF oxidation consisting of 469 species and 1221 reactions,and they performed the validated experiment in a variable pressure flow reactor at 1.27 MPa.Andrae et al.9added the toluene sub-mechanism to the PRF detailed chemical kinetic mechanism and developed a detailed chemical kinetics model(1083 species and 4635 reactions)for the autoignition of TRF oxidation,which was validated using shock tube autoignition delay time data. Afterwards,they presented a semidetailed mechanism10(137 species and 633 reactions)in a HCCI engine on the autoignition of TRF.Sakai et al.11mechanisms for iso-octane and n-heptane were added to a detailed toluene submechansim.The model showed generally good agreement with ignition delay times measured in a shock tube and a rapid compression machine. Sakai et al.proposed a toluene sub-mechanism started from the toluene mechanism developed by Pitz et al.,12which was merged to the previous PRF model.The TRF oxidation mechanism(783 species and 2883 reactions)was validated by existing shock tube and flow tube data.Anderlohr et al.13proposed a TRF oxidation mechanism(536 species and 3000 reactions) which was based on the PRF autoignition model coupled with the model for the oxidation of toluene.Flow rate and sensitivity analysis were performed in order to explain the low temperature chemical kinetics,especially the impact of NOxon hydrocarbon oxidation.
At present,most of TRF mechanisms are the detailed chemical kinetic model,and the development of TRF oxidation mechanisms is still in primary stage.The detailed chemical kinetic model is large and difficult to use in engine models.It spends too much money and time to apply the 3D-CFD combustion simulation with the detailed mechanisms.In sum,the research on the TRF oxidation mechanism is not enough,specially the reduced mechansim.The necessity of developing the reduced TRF mechanism is brought out.In this study,a new reduced mechanism is suitable for generalized numerical simulation which developed and validated by different experiments related to HCCI combustion.
2 Construction of TRF kinetic model
Because this mechanism is mainly for HCCI engine application,it is used over the range of low equivalence ratio and high pressure.Since the ignition of HCCI engine is very sensitive to temperature,the mechanism should include both low temperature and high temperature kinetics.
The reaction mechanism steps of iso-octane and n-heptane oxidation are similar.However,the rate coefficients for many of the individual reactions are different reflecting the very different structure of these two molecules.In following reactions RH denotes paraffins,and Q denotes olefins structure.A chemical kinetic mechanism proposed by Tanaka et al.3is used as a starting model,here the low temperature mechanism includes the following reactions.
The mechanism in the case of iso-octane is as follows.Fuel oxidation is initiated to form the alkyl radical R and HO2when H atoms are abstracted by oxygen molecules RH+O2⇌R+HO2(Reaction(R1,R14)).At low temperature,in the sequence of reactions:an alkyl radical is oxidized by an oxygen molecule R+O2⇌RO2(the first oxygen addition process,Reaction(R2, R15)),an internal hydrogen is rearranged RO2⇌QOOH(Reaction(R3,R16)),another oxygen molecule is added to a hydroperoxy alkyl radical QOOH+OB2⇌OOQOOH(the second oxygen addition process,Reaction(R4,R17)),and the product decomposes irreversibly OOQOOH➝OH+OROOH(Reaction (R5,R18)),as a result,an OH radical and an ketohydroperoxide are formed.After the reaction of an OH radical with a fuel molecule RH+OH➝R+H2O(Reaction(R6,R19)),highly exothermic chain-cycle reactions from reaction(R2)to(R6)are subsequently completed.Then the branching reaction started, lots of heat is released and the temperature rises rapidly,until the following competing reactions R+O2⇌Q+HOR2(R7,R20) becomes faster than reaction(R2).
Tanaka et al.3included three global reactions and directly decomposed OC8H15O and OC7H13O into CO and H2O.Because the presence of the important intermediate product is ignored, it is difficult for Tanaka model to predict HC and CO emissions.At the same time,their sensitivity analysis shows that these reaction paths are relatively unimportant to ignition delay times and heat release ratio using the species OC8H15O and OC7H13O.Tsurushima14pointed out that CH2O and HO2were both key intermediate species,and the fuel consumption rate during the low-temperature chain reaction was too small with Tanaka model.In order to raise prediction level,the following several reactions are included and the large molecules formed during the low temperature stage accordingly break up into smaller molecules.In these reactions,R′denotes paraffins with C5or C6.The decomposition process of ketohydroperoxide,the reaction OROOH➝R′CO+CH2O+OH(R8,R21)and R′CO+O2➝small molecule+CO+HO2(R9,R22)were modified.The reaction Q+O2➝R′+CH2+HCO(R10,R23)was added to condider CH2O formation.Then the reaction RH+HO2⇌R+H2O2(R11,R24)was added to maintain the balance of production and consumption of alkyl radical.In this model,betascission of alkyl radicals is considered in the reaction R➝R′+ small molecules(R12,R25).Afterwards,smaller alkyl radical R′decomposes to small molecules through the reaction R′⇌small molecules(R13,R26).

Fig.1 Major reaction branches of toluene oxidation
From the existing mechanisms of the toluene,similarity in reaction pathways can be noted.The overall reaction proceeds primarily through C6H5CH3→C6H5CH2→C6H5CHO→C6H5CO→C6H5→C6H5O→C6H5OH,followed by the ring-breaking reactions.Fig.1 presents the chemical reaction paths of toluene. The initial mechanism of the toluene oxidation is from Sivara-makrishnan et al.4Since benzene is a key intermediate in the oxidation of toluene,the benzene submodel should be considered.Selected benzene reactions were mainly based on Andrae et al.10from Alzueta model,15which was validated by comparison with the experimental data as well as flow reactor and mixed reactor data from literature.The validated experiments were also performed under plug-flow conditions,at excess air ratios ranging from close to stoichiometric to very lean,which are relevant to HCCI combustion.Some additional reactions including the species CH2OH,CH2CO were added,based on the work by UCSD.16The toluene sub-mechanism is shown in Appendix(available free of charge via the internet at http://www. whxb.pku.edu.cn).
Some rate constantsare updated mainly fortoluene (C6H5CH3)and benzyl radical(C6H5CH2)reactions taken from recent studies.The rate coefficients of the toluene reactions are derived from published studies of Oehlschlaeger et al.17-19and Seta et al.20Oehlschlaeger et al.investigated some reactions of toluene using ultraviolet laser absorption of benzyl radicals in shock tube experiments.The rate coefficient results were in excellent agreement with the previous rate determinations.They thought the reaction of toluene with H-atoms producing benzyl radicals and molecular hydrogen is an important elementary reaction in the toluene pyrolysis and oxidation reaction systems. The reaction retards ignition during toluene oxidation by removing an H-atom that otherwise would lead to chain branching via reaction with O2.The decomposition of toluene is an important initiation step in the oxidation of toluene at moderateto high-temperatures(>1200 K),and the toluene decomposition takes place via two channels:C6H5CH3⇌C6H5CH2+H (R27)and C6H5CH3⇌C6H5+CH3(R28).The reaction of toluene with molecular oxygen is also the primary initiation reaction during the oxidation of toluene,which has been studied.Seta et al.measured the overall rate constants of the toluene reaction with OH radical and estimated channel specific rate coefficients of this reaction on the basis of RRKM-master equation analysis.This reaction is seen to account for the major consumption of toluene during the induction period.
Benzyl is a resonantly stabilized radical that commonly occurs as an intermediate in the oxidation and pyrolysis of toluene.Benzyl is generated as an intermediate at significant concentrations during the oxidation of toluene via the reaction of toluene with molecular oxygen,the reaction of toluene with radical species,and the toluene thermal decomposition channel leading to benzyl radicals and H-atoms.Silva et al.21pointed out that the bimolecular reactions of benzyl with HO2were important in the oxidation of toluene,especially at low to moderate temperatures.We also updated rate constants of the benzyl radical reactions according to the studies of Andrae et al.10In simulation for the experiment of Davidson et al.,22the reaction C6H5CH2OO⇌C6H5CHO+OH(R48)was found to be very sensitive to the overall reactivity for neat toluene-air mixtures and the rate constant was tuned for best fit of model to the shock tube data that proposed by Andrae et al.,10and Klotz et al.23pointed out that considering the formation of 1,3-butadiene from toluene was necessary and the addition of iC4H5reactions significantly improved predictions of 1,3-butadiene and acetylene.Seven reactions about iC4H5were added into the toluene submechanism.Andrae et al.10performed simulations with and without the reactions to checked the importance of cross reactions.The introduction of cross reactions into a detailed model for TRF was found to improve the results in those particular HCCI simulations.In this work,five cross reactions (R147-R151)were merged to the TRF mechanism based on previous work of Andrae.Cross reactions between phenyl radicals and the primary reference fuels are also of importance, which were considered in the new model.
Unlike previous TRF mechanisms incorporating detailedmechanism,in order to keep the mechanism size compact,thereactions are taken from a reduced mechanism for HCCI engines developed by Patel et al.24.The main branching reaction is H2O2+M⇌OH+OH+M(R173)at the temperatures from 900 to 1100 K,and the reaction H+O2+M⇌O+OH+ M(R174)is the significant branching reaction at higher temperatures,when H radical becomes an important species.It may be noted,the role of the competition between this branching reaction(R174)and the recombination process reaction H+ O2+M⇌HO2+M(R175)cannot be disregarded.Reaction (R174)is added to the reactions of C1-C3oxidation developed by Patel.Then,the whole toluene reference fuels oxidation mechanism is completed.The final version of the TRF mechanism consists of 70 species and 196 reactions.
3 Model validation
3.1 Validation of ignition delay
A parameter often used in autoignition studies is the ignition delay,which is the time required for autoignition to happen once the fuel-air mixture is raised to a given pressure and temperature and held at that condition in a shock tube or a rapid compression machine.We have compared the model predictions with some representative experimental results.All calculations were performed by using CHEMKIN 4.1 program package,25and an adiabatic homogeneous reactor is assumed for simulations of ignition delay times.All the figures of ignition delay are shown in semi-log plot.In terms of detailed studies, the whole validation can be divided into three categories:primary reference fuels,toluene,and toluene reference fuels.
3.1.1 Primary reference fuels
Mechanism validation of primary reference fuels were performed for the ignition delay times from the shock tube experiments in previous studies,26,27The ignition delay time is defined in the experiment as the time interval between the arrival of the reflected shockwave and the onset of ignition in combustion chamber.And computation was done under completely homogeneous,constant volume,and adiabatic conditions.
Fig.2(a)is the comparison of ignition delays between the cal-culation and the shock tube experiments of Fieweger et al.26at initial pressures of 1.3,3.4,4.5 MPa with stoichiometric iso-octane-air mixture.At the higher pressures,the ignition delay time becomes shorter,but the slope of the curves is essentially unchanged.Fig.2(b)is computed ignition delays of iso-octane for the different equivalence ratios at a pressure of 4 MPa also compared to experimental data.The ignition delay times increase with decreasing equivalence ratio.The values of ignition delay show a pronounced negative temperature coefficient (NTC)behaviour for stoichiometric mixtures and rich mixtures.The ignition delay with the new model seems good agreements with the experiments.In Fig.3 a comparison of ignition delay times for stoichiometric fuel/air mixture with different octane numbers at an initial pressure of 4 MPa with comparisons to the shock tube data of Fieweger.For temperatures above 1000 K,there is no significant difference among the ignition delay times of the mixtures.Overall,however,the ignition timing advances with the decrease of the octane number.In this graph,the sensitivity of ignition delay to the octane number shows excellent agreement to the shock tube data.
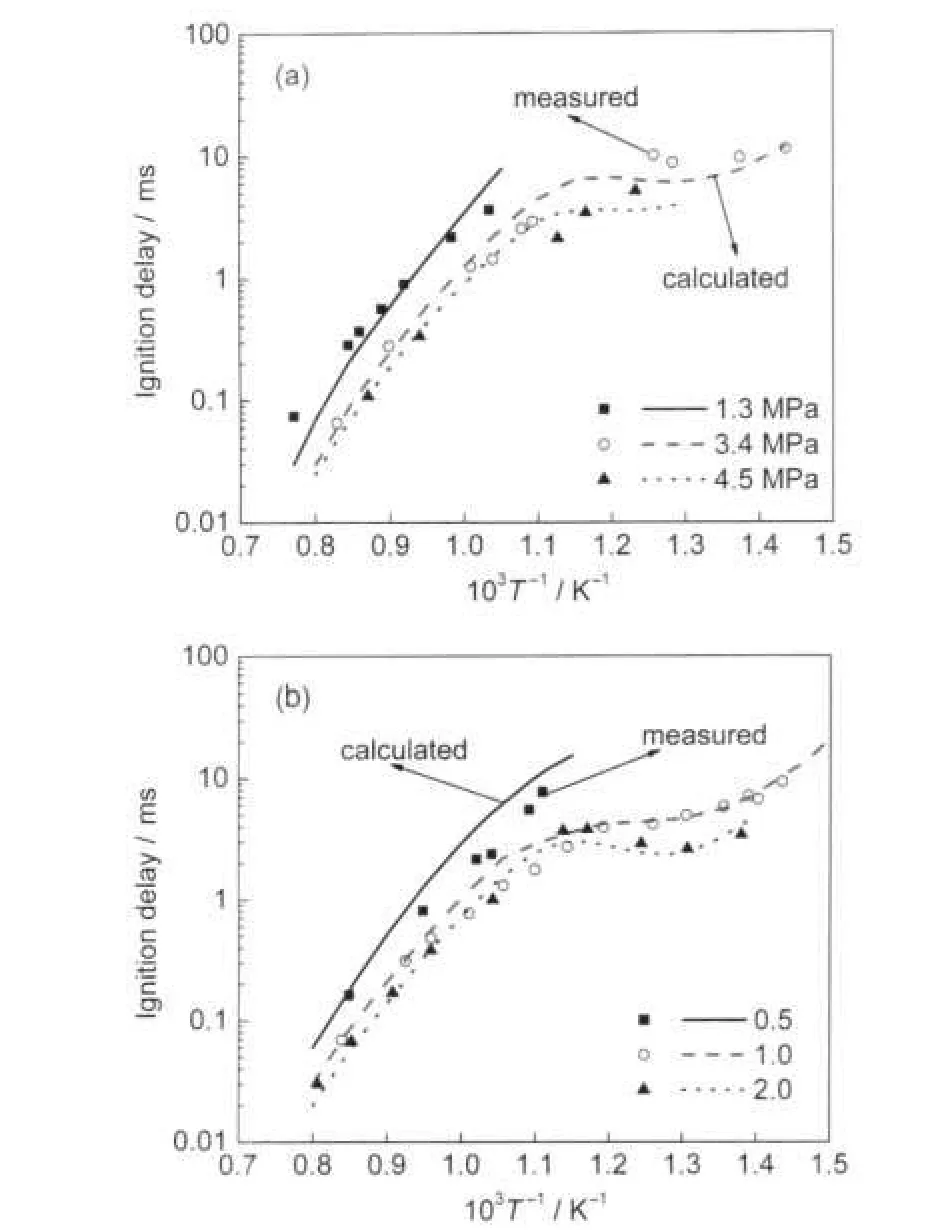
Fig.2 Comparison of experimental26and computed ignition delay of iso-octane-air mixture at different pressures(a)and equivalence ratios(b)
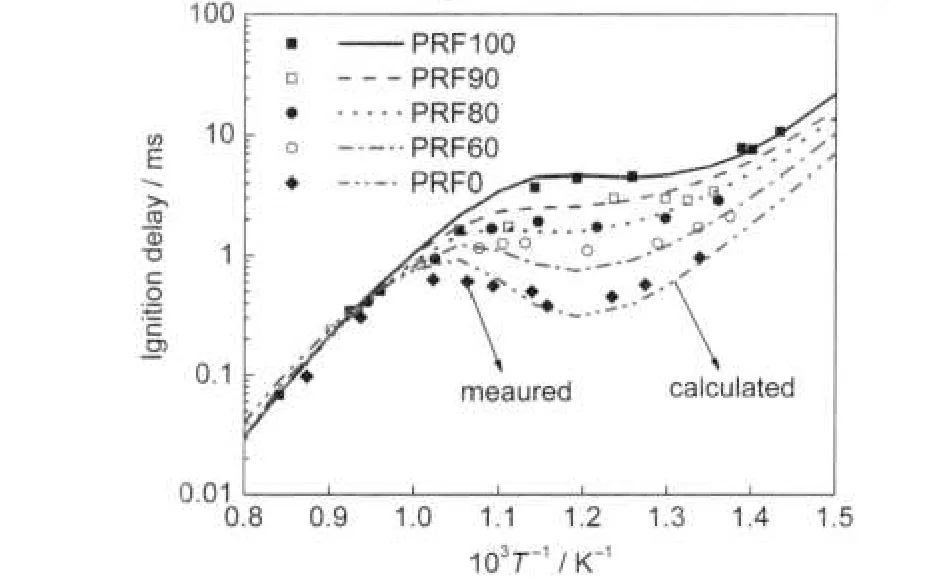
Fig.3 Comparison of experimental26and calculated ignition delay of PRF-air mixture
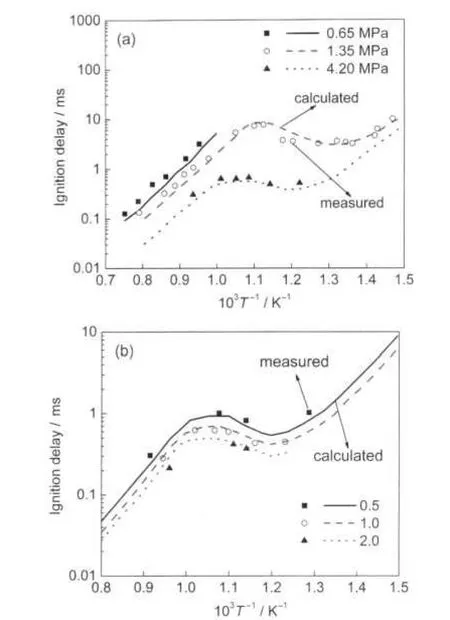
Fig.4 Comparison of experimental27and calculated ignition delay of n-heptane-air mixture at different pressures(a)and equivalence ratios(b)
Fig.4(a)shows computed ignition delays of stoichiometric n-heptane/air mixture at initial pressures of 0.65,1.35,4.2 MPa,with comparison to shock tube data of Ciezki et al.27Fig.4 (b)gives computed ignition delays of n-heptane at a pressure of 4 MPa and equivalence ratios of 0.5,1.0,and 2.0.In these figures,it can be seen that the ignition delays of PRF with new model shows very good agreement with the experimental data. The calculation is sensitive to changes in temperature and mixture composition and captures the NTC region.
3.1.2 Toluene
Fig.5(a)shows the comparison between the calculated results and the experimental results measured by Davidson et al.22in shock tube for toluene.The ignition delay time is defined in the experiment as the time interval between the arrival of the reflected shockwave and the onset of ignition at the sidewall observation location.The arrival of the reflected shockwave was determined by the step rise in pressure,and the onset of ignition was determined by monitoring either the pressure history or the emitted light corresponding to an intermediate species.The onset of ignition from the pressure history as well as both CH*and OH*emission were defined by locating the time of steepest rise and linearly extrapolating back in time to the pre-ignition baseline.As can be seen from Fig.5(a),good agreement between experimental and calculated data is obtained at a pressure of 5 MPa,especially for the conditions in relative high temperature regime.But the calculation tends to predict longer ignition delays than the experimental data in relative low temperature regimes.The calculation over-predicts the ignition delay times below 1250 K for φ=0.5 and below 1080 K for φ=1.0.
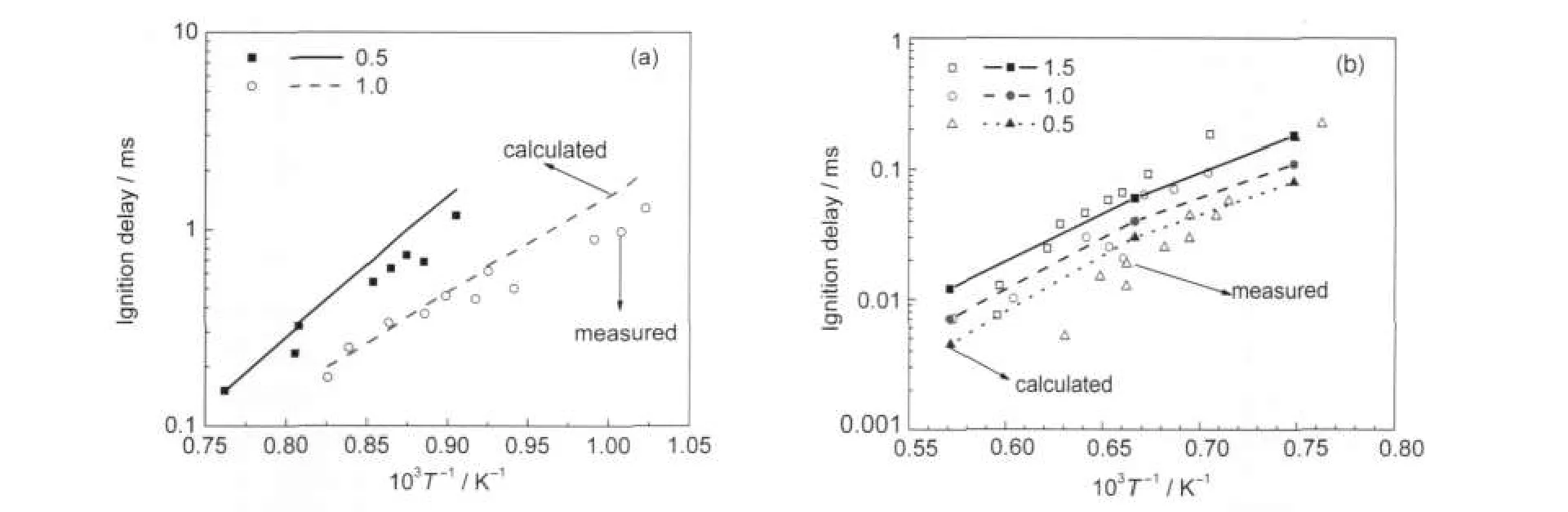
Fig.5 Comparison of experimental[Davidson et al.22(a),Pitz et al.12(b)]and calculated ignition delay of toluene-air mixture
In Pitz's shock tube experiment,1210%of the maximum OH emission was used as an indication of ignition.The toluene concentration in the reactants kept constant at 1.25%,and the values of equivalence ratio were 0.5,1.0,and 1.5.The reflected shock pressures and temperatures ranged from 8.0 to 9.4 MPa and 1300 to 1900 K,respectively.Using the new reduced TRF mechanism,simulation was carried out in accordance with Pitz's experimental conditions.Fig.5(b)presents the comparison of the computed ignition delays and the measuresed ones for the different equivalence ratios.As a whole,the predictions are satisfactory,and the trends of the calculated curves are in agreement with the experiment.
3.1.3 Toluene reference fuels
In Gauthier's shock tube study,28the two ternary surrogates used in the ignition delay time experiments consisted of iso-octane,toluene,and n-heptane in the following proportions:surrogateAand surrogate B.Fuels and their properties are presented in Table 1.Ignition delay times of toluene reference fuels were measured at temperatures 850 to 1200 K.The definition of ignition delay time is the same as Davidson et al.22(see Section 3.1.2).Fig.6 shows a comparison of ignition delay times for stoichiometric fuel-air mixture with comparisons to the shock tube data.At this temperature range and higher pressure conditions,the variation range of TRF ignition delay time is narrower than that of PRF in Fig.3.The computed ignition delay with new model shows good agreement with the experimental data.
3.2 Validation in HCCI engines
A single zone model is used in the computation assuming that temperature,pressure and species concentrations are uniform in the chamber.The wall heat transfer,residual gasesfrom previous cycle,and the temperature inhomogeneity in the boundary and crevice zones are ignored in the model.Despite its simplicity,the single zone model is a useful tool for investigating certain fundamental aspects of HCCI combustion.The operating conditions of each engine experiment in the simulation are given in Table 2.
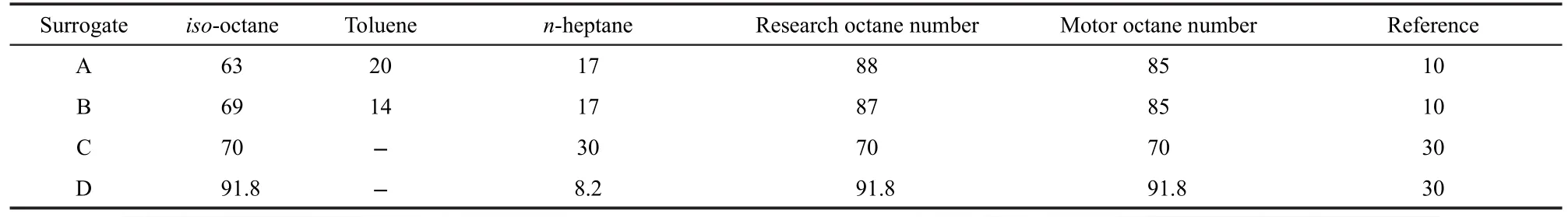
Table 1 Compostion(liquid volume fraction(%))of TRF investigated
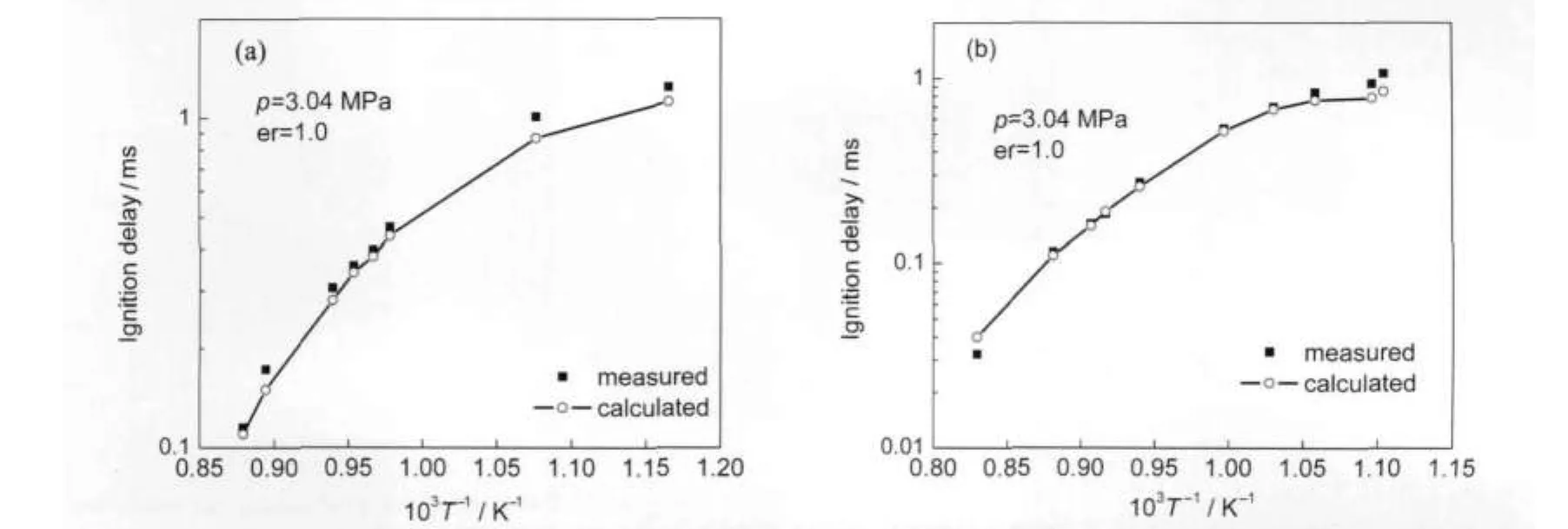
Fig.6 Comparison of experimental28and calculated ignition delay of TRF-air mixture(a)surrogateA,(b)surrogate B;er=equivalence ratio
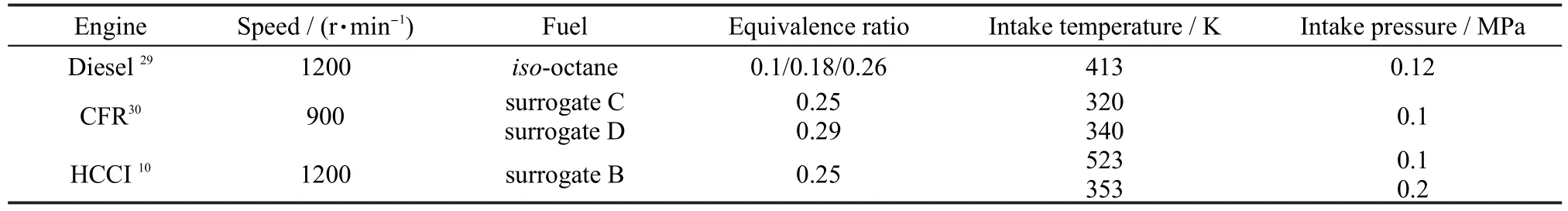
Table 2 Engine operating conditions
Dec et al.29have performed a series of experiments using iso-octane as fuel in HCCI engine.Fig.7 shows the cylinder pressure profiles of modeling and experiment for various equivalence ratios at the compression ratio of 18,engine speed of 1200 r·min-1,and initial pressure of 0.12 MPa.Other operating parameters were held constant.The highest pressure increases with the increases of fuel load.Because of the assumption of adiabatic condition in the single zone model,the maximum pressures are over predicted,but the trends of the calculation results are in accordance with experiment results for three loads.
HCCI combustion data30for PRF fuel-air mixtures tested in a single-cylinder CFR research engine were used to validate the predictions of PRF autoignition under engine conditions. The compression ratio and the engine speed were set at 16.55, and 900 r·min-1respectively for all the cases.The intake pressure was maintained at 0.1 MPa.The results of the fuel/air mixture(surrogate C and surrogate D)HCCI experiments are shown in Fig.8,and fuels and their properties are presented in Table 1.The cylinder pressure histories for surrogate C at the initial temperature of 320 K and the equivalence ratio of 0.25 are shown in Fig.8(a).Surrogate D at the initial temperature of 340 K and the equivalence ratio of 0.29 shown in Fig.8(b).Curran et al.6,7developed detailed reaction mechanism for iso-octane and n-heptane,which were widespreadly applied.The reduced mechanism and detailed mechanism are compared by computation.To be noted,the pressures predicted by the present reduced mechanism are almost the same as that by the PRF detailed mechanism.However,under the computing environment of 2.51 GHz CPU and 2 GB DDR,it only took 2 s on average with the reduced mechanism,which is far less than the time 420 seconds with the detailed mechanism.
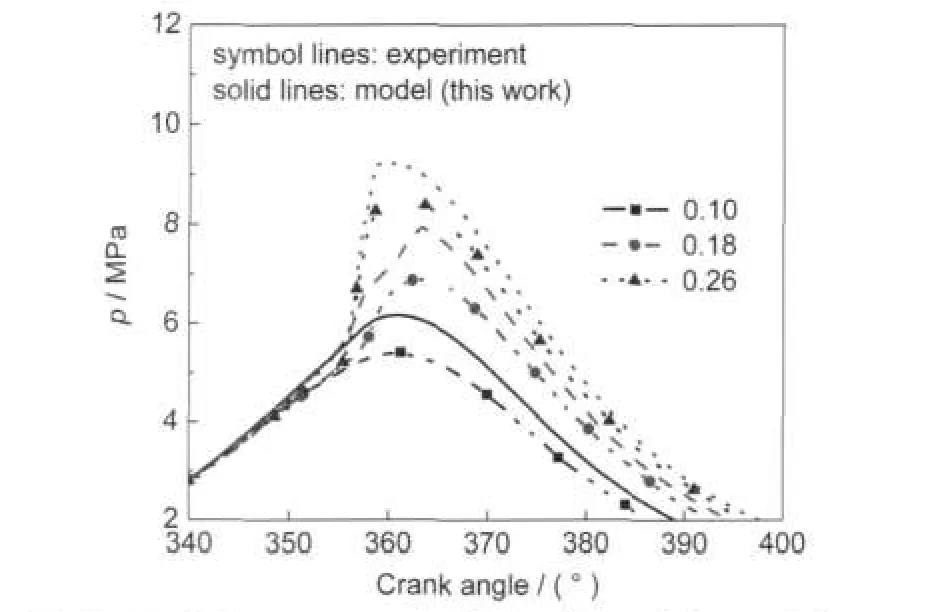
Fig.7 Cylinder pressure profiles for the modeling and the experiment29with iso-octane at different aquivalence ratios
HCCI experiments have been conducted with TRF in the HCCI engine by Andrae et al.10Fig.9 shows both the experimental results and the simulation results with surrogate B(see Table 1)at the engine speed of 1200 r·min-1and the compression ratio of 14.04.The start of calculations is at 99 degree before top dead center.Two different initial conditions are set: the first condition is at the initial temperature of 489 K and initial pressure of 0.15 MPa(Fig.9(a));the second condition is at the initial temperature of 435 K and initial pressure of 0.33 MPa(Fig.9(b)).The results show good agreements both at the two conditions.Fig.9 also shows heat production predicted by the model.Maximum heat production of two operating conditions is at the crank angle of-1.8 and 1.8 degree after top dead center,respectively.The ignition delay time is more advanced at condition 1(high temperature and low pressure)compared with condition 2(low temperature and high pressure).However,maximum heat production of condition 2 is higher.

Fig.8 Cylinder pressure profiles for the modeling and the experiment with PRF(a)surrogate C,(b)surrogate D
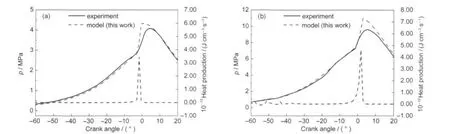
Fig.9 Cylinder pressure profiles for the simulation and the experiment10with TRFcondition:(a)high temperature and low pressure;(b)low temperature and high pressure
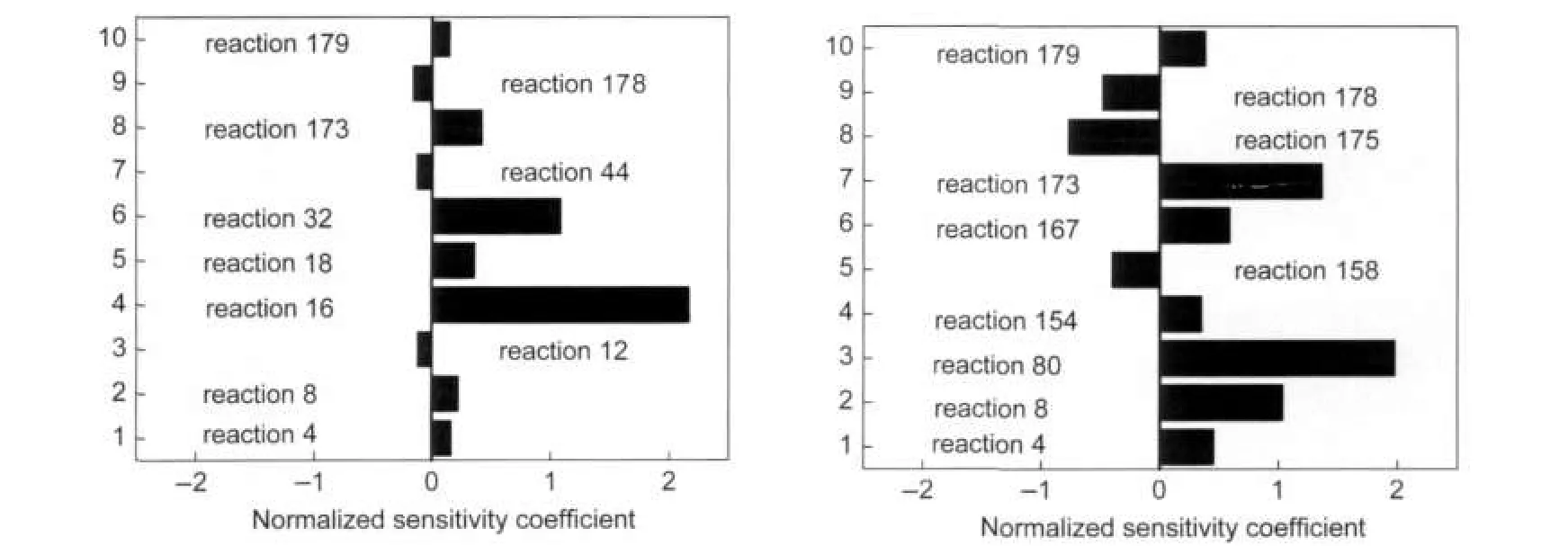
Fig.10 Normalized sensitivity coefficients for TRF at different conditionscondition:(a)high temperature and low pressure;(b)low temperature and high pressure
Fig.10 presents a bar plot of normalized sensitivity coefficients for surrogate B.The sensitivity calculation was done for the 10 most sensitive reactions at the crank angle for maximum heat production.The conditions for the calculations are conditions 1 and 2 as in Fig.9.For condition 1(see Fig.9(a)),the reaction with two of the highest positive sensitivity are alkyl radical isomerization in C7H15OO⇌C7H14OOH(R16)and the toluene reaction with O radical in C6H5CH3+O=C6H5CH2+OH (R32).
For the case of condition 2(see Fig.9(b)),on the other hand, reactions(R16)and(R32)are not among the 10 most sensitive reactions.Instead,the reaction(R80)of phenol radicals with O2has the highest positive sensitivity.The sensitivity results for higher pressure are the same as shock tube experiments performed by Sivaramakrishnan et al.4

For both operating conditions,the following five reactions show higher sensitivity.
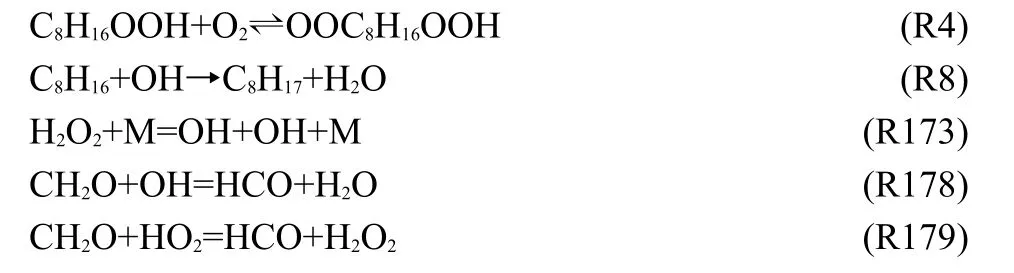
Although there are some small variations,the trend in sensitivity to reactions is the same with changing operating conditions.Reaction(R173)initializes the thermal explosion,and the reaction of fuel oxidation with the product OH radicals happens immediately,which causes the temperature rise rapidly. Fig.10 shows that the reactions(R4)and(R8)have high positive sensitivity,especially in the condition of low temperature and high pressure.The reaction(R178)of formaldehyde (CH2O)with OH radicals shows a higher negative sensitivity, and the reaction(R179)of CH2O with HO2radicals also shows a higher positive sensitivity.Formaldehyde is very important intermediate,which should not be neglected in constructing mechanism.
4 Conclusions
(1)A reduced toluene reference fuel(TRF)oxidation mechanism for HCCI combustion comprised of 70 species and 196 reactions was developed.The mechanism has been validated by comparing computed results with experimental data within the HCCI combustion mode.
(2)In shock tube conditions,computed ignition delay time with the new TRF mechanism was validated with the experimental data in literature with iso-octane,n-heptane,and toluene as fuels at different conditions.Good agreements of ignition delay times are obtained.
(3)In HCCI engine conditions,the comparison of the computed and measured results with PRF/TRF as fuels indicates that the combustion process can be well predicted with this mechanism.A sensitivity analysis from low intake pressureor high intake temperature to high intake pressure or low intake temperature shows that reaction(R80)of phenol radicals with O2has the higher sensitivity as the pressure rising,and formaldehyde(CH2O)is very important intermediate,which should not be neglected.
(4)As a conclusion,the new TRF model is believed to have enough accuracy for the HCCI combustion simulation.It is suitable for toluene/PRF/TRF as fuels.And the present reduced model can be a useful tool to study HCCI engine operation.
(1)Yao,M.F.;Zheng,Z.L.;Liu,H.F.Prog.Energ.Combust. 2009,35,398.
(2)Westbrook,C.K.Proc.Combust.Inst.2000,28,1563.
(3)Tanaka,S.;Ayala,F.;Keck,J.C.Combust.Flame 2003,134, 219.
(4) Sivaramakrishnan,R.;Tranter,R.S.;Brezinsky,K.Proc. Combust.Inst.2005,30,1165.
(5) Pitz,W.J.;Cernansky,N.P.;Dryer,F.L.;Egolfopoulos,F.N.; Farrell,J.T.;Friend,D.G.;Pitsch,H.SAE Tech.Pap.Ser.2007, 2007-01-0175.
(6) Curran,H.J.;Gaffuri,P.;Pitz,W.J.;Westbrook,C.K.Combust. Flame 1998,114,149.
(7) Curran,H.J.;Gaffuri,P.;Pitz,W.J.;Westbrook,C.K.Combust. Flame 2002,129,253.
(8) Chaos,M.;Zhao,Z.;Kazakov,A.;Gokulakrishnan,P.; Angioletti,M.;Dryer,F.L.APRF+Toluene Surrogate Fuel Model for Simulating Gasoline Kinetics.In 5th U.S. Combustion Meeting,March 25-28,2007;University of California,San Diego,California,Paper#E26.
(9)Andrae,J.C.G.;Björnbom,P.;Cracknell,R.F.;Kalghatgi,G. T.Combust.Flame 2007,149,2.
(10)Andrae,J.C.G.;Brinck,T.;Kalghatgi,G.T.Combust.Flame 2008,155,696.
(11) Sakai,Y.;Miyoshi,A.;Koshi,M.;Pitz,W.J.Proc.Combust. Inst.2009,32,411.
(12) Pitz,W.J.;Seiser,R.;Bozzelli,J.W.;Seshadri,K.;Chen,C.J.; Costa,D.;Fournet,R.;Billaud,F.;Battin-Leclerc,F.; Weatbrook,C.K.Chemical Kinetic Study of Toluene Oxidation.In 29th International Symposium on Combustion, Hokkaido University,Sapporo,Japan,July 21-26,2002; Elsevier:New York,2002,UCRL-JC-125890.
(13)Anderlohr,J.M.;Bounaceur,R.;Pires Da Cruz,A.; Battin-Leclerc,F.Combust.Flame 2009,156,505.
(14) Tsurushima,T.Proc.Combust.Inst.2009,32,2835.
(15)Alzueta,M.U.;Glarborg,P.;Dam-Johansen,K.Int.J.Chem. Kinet.2000,32,498.
(16)Welcome to Chemical-Kinetic Mechanisms for Combustion Applications.http://maeweb.ucsd.edu/~combustion/cermech/ index.html(accessedAug 27,2010).
(17) Oehlschlaeger,M.A.;Davidson,D.F.;Hanson,R.K.Proc. Combust.Inst.2007,31,211.
(18) Oehlschlaeger,M.A.;Davidson,D.F.;Hanson,R.K.J.Phys. Chem.A 2006,110,9867.
(19)Oehlschlaeger,M.A.;Davidson,D.F.;Hanson,R.K.Combust. Flame 2006,147,195.
(20) Seta,T.;Nakajima,M.;Miyoshi,J.Phys.Chem.A 2006,110, 5081.
(21) Silva,G.;Bozzelli,J.W.Proc.Combust.Inst.2009,32,287.
(22)Davidson,D.F.;Gauthier,B.M.;Hanson,R.K.Proc.Combust. Inst.2005,30,1175.
(23) Klotz,S.D.;Brezinsky,K.;Glassman,I.Modeling the Combustion of Toluene-Butane Blends.In 27th Symposium (International)on Combustion,University of Colorado at boulder,USA;The Combustion Institute:Pittsburgh,PA,1998; 337.
(24) Patel,A.;Kong,S.C.;Reitz,R.D.SAE Tech.Pap.Ser.2004, 2004-01-0558.
(25)Kee,R.J.;Rupley,F.M.;Miller,J.A.;et al.CHEMKIN Release 4.1,Reaction Design:San Diego,CA,2006.
(26)Fieweger,K.;Blumenthal,K.R.;Adomeit,G.Combust.Flame 1997,109,599.
(27)Ciezki,H.K.;Adomeit,G.Combust.Flame 1993,93,421.
(28) Gauthier,B.M.;Davidson,D.F.;Hanson,R.K.Combust. Flame 2004,139,300.
(29) Dec,J.E.;Sjöberg,M.SAE Tech.Pap.Ser.2003,2003-01-0752.
(30)Aroonsrisopon,T.;Sohm,V.;Werner,P.;Foster,D.E.; Morikawa,T.;Lida,M.SAE Tech.Pap.Ser.2002, 2002-01-2830.
猜你喜欢
今日农业(2021年14期)2021-10-14
中华养生保健(2020年9期)2021-01-18
世界有色金属(2019年9期)2019-07-03
无机化学学报(2019年2期)2019-02-27
大东方(2018年9期)2018-10-21
红岩春秋(2018年6期)2018-06-28
人大建设(2018年3期)2018-06-06
信息记录材料(2016年4期)2016-03-11
图书馆学刊(2015年8期)2015-12-26
应用化工(2014年7期)2014-08-09
