
高脂饮食导致大鼠肝脏胰岛素抵抗的作用机制研究*
2011-11-22 07:02李兰芳唐国涛喻翠云陈临溪
中国病理生理杂志 2011年2期
李兰芳, 郭 玉, 唐国涛, 曹 轩, 喻翠云, 陈临溪
(南华大学药物药理研究所,湖南 衡阳 421001)
高脂饮食导致大鼠肝脏胰岛素抵抗的作用机制研究*
李兰芳, 郭 玉, 唐国涛, 曹 轩, 喻翠云, 陈临溪△
(南华大学药物药理研究所,湖南 衡阳 421001)
目的本研究旨在建立SD大鼠胰岛素抵抗模型,观察高脂饲料喂养的SD大鼠肝脏中氧化应激以及胰岛素抵抗的发生,分析胰岛素抵抗状态下活性氧(ROS)的变化,初步探讨ROS的主要来源。方法以高脂饲料喂养6只4周龄雄性SD大鼠12周,建立大鼠胰岛素抵抗模型。用优越血糖仪以电子感应法测定血糖,放射免疫法检测血清胰岛素水平。二氢乙啶(DHE)染色观察肝脏组织中的ROS水平。Western blotting检测NADPH氧化酶3(NOX3)的表达。结果以高脂饲料喂养12周后,大鼠空腹血糖水平略有上升,但与对照组的大鼠相比无显著差异,而胰岛素敏感指数降低。蒽酮法的检测结果显示高脂饲料喂养大鼠肝组织糖原含量显著降低,高脂饮食大鼠肝组织中NOX3的表达显著增加,DHE染色显示肝组织ROS水平显著增加,提示ROS在肝胰岛素抵抗发生中起重要作用。结论高脂饲料喂养SD大鼠胰岛素敏感指数降低,肝组织中NOX3表达和ROS水平显著增加,糖原含量显著降低。
胰岛素抵抗; NADPH氧化酶; 氧化性应激
糖尿病是以血浆中葡萄糖(简称血糖)水平升高为主要特征的代谢性疾病。而引起血糖升高的病理生理机制主要是胰岛素分泌缺陷和(或)胰岛素作用缺陷(胰岛素抵抗)。胰岛素抵抗是由遗传和环境因素导致的,表现为机体对胰岛素生理作用的反应性降低,即胰岛素促进葡萄糖摄取作用受损,导致代偿性胰岛素分泌增多,其重要标志为高胰岛素血症,具体表现为外周组织对胰岛素敏感性下降,以及对葡萄糖的利用障碍。胰岛素抵抗主要发生在脂肪、肝脏和骨骼肌,因为这些组织的细胞含有大量的胰岛素受体,它们能结合胰岛素从而在调节葡萄糖代谢稳态上发挥重要作用。
在胰岛素抵抗发生的机制中,氧化应激被认为是其中最为重要的机制。2004 年在美国糖尿病学会年会上就有学者提出了胰岛素抵抗的统一发病机制,认为共同基础就是氧化应激[1]。2004年欧洲糖尿病研究学会也提出共同土壤学说,认为胰岛素抵抗、糖尿病和心血管疾病都有共同发生机制,即氧化应激。氧化应激是上述疾病发生的共同土壤。氧化应激在胰岛素抵抗中的作用以及抗氧化的治疗已成为胰岛素抵抗和2型糖尿病防治的重要研究方向[2]。虽然研究已证实氧化应激在胰岛素抵抗发生中起着重要的作用,但是氧化应激在肝脏胰岛素抵抗中的具体作用机制还有很多问题有待更进一步地深入研究和探讨。本研究旨在通过喂养高脂饲料,建立SD大鼠胰岛素抵抗模型。观察肝脏胰岛素抵抗状态下氧化应激的水平,分析胰岛素抵抗状态下活性氧(reactive oxygen species, ROS)的变化,明确ROS的来源,初步探讨NADPH氧化酶3(NOX3)在肝细胞胰岛素抵抗中的作用。
材 料 和 方 法
1材料
肌、肝糖原检测试剂盒(南京建成生物试剂公司),活性氧检测试剂盒(碧云天),Electro-chemical luminescence 免疫印迹试剂盒(Amersham Pharmacia)[125I] radioimmunoassay kit(Insulin)(Linco),dihydroethidium(DHE,Sigma),羊抗NOX 3抗体(A-12)(SC-34699) (Santa Cruz),actin(I-19)(sc-1616) (Santa Cruz),兔抗山羊IgG(H+L)(北京中杉金桥)。
2实验方法
2.1实验动物分组 高糖高脂喂养4周龄雄性Sprague Dawley(SD)大鼠(由北京大学医学部实验动物中心提供)以建立胰岛素抵抗动物模型。将大鼠随机分为2组:正常对照组(6只)给予普通饲料喂养;高脂喂养大鼠(6只)给予的饲料为普通饲料中添加20%脂肪(猪油和蛋黄粉等份)和20%蔗糖。喂养12周后,称体重后断尾法留取空腹血标本,取血测定空腹血糖浓度和血清胰岛素水平,并计算胰岛素敏感指数。
2.2血清标本的采集 断尾法留取空腹血标本,离心后分离血清,将血清样品放入-20 ℃低温冰箱中保存,供以后测定时使用。
2.3肝脏组织标本的采集 将大鼠用戊巴比妥钠麻醉后开始手术。采用腹中切口进入腹腔,充分暴露手术视野。将获取的肝脏组织标本快速分别放入液氮中冷冻和4%甲醛液中固定。所有操作完成后将大鼠在麻醉状态下采用气胸法处死。
2.4指标测定
①血糖 采用电子感应法,用罗氏公司(Roche)的优越血糖仪(ACCU-CHEK ADVANTAGE)测定。
②血清胰岛素 采用放射免疫法测定(用大鼠标准品绘制标准曲线,批内变异<4%,批间变异<8%)。所有标本同一批测定完成。
胰岛素敏感指数计算(insulin sensitivity index, ISI):测定空腹血糖和空腹血清胰岛素后,按照公式 ln[1/(空腹血糖 × 空腹血清胰岛素)]计算。
③肝组织糖原含量测定 按照肌、肝糖原检测试剂盒的说明操作。样本处理:收集细胞,称重。按样本重量(mg):碱液体积(μL)=1∶3,一起加入试管中,沸水煮20 min,流水冷却。将糖原水解液进一步制备成糖原检测液:肝糖原检测液为1%,加蒸馏水的量为:细胞重量×100-细胞重量×4=细胞重量×96,混匀。按试剂盒说明将糖原检测液与显色剂混合,混匀后置沸水中煮5 min,冷却后于620 nm波长,1 cm光径,空白管调零,测各管A值,计算糖原含量。
④肝组织内ROS水平测定 取出液氮冻存的肝脏组织至-20 ℃平衡温度后,OCT包埋制备成冰冻块,用冰冻切片机以5 μm厚度连续切片。DHE原液用PBS按1∶1 000稀释即为工作液,将冲洗切片用的PBS预冷。用PBS冲洗冰冻切片2-3次后,将DHE工作液滴加于冰冻切片上,室温避光孵育15 min。PBS洗片,2 min×3次。50%甘油封片后荧光显微镜下观察。
3统计学处理
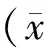
结 果
1高脂喂养大鼠体重变化
高脂喂养12周后称体重(body weight, BW),普通饲料喂养大鼠的BW为(576.33±29.36)g,而高脂喂养大鼠的BW为(602.43±46.18)g,体重比对照组略有增加但无显著差异,见图1。

Figure 1. The body weight of rats fed with high-fat diet(HF)..
2高脂喂养大鼠空腹血糖浓度变化
采用电子感应法,测定大鼠空腹血糖浓度(fasting blood glucose, FBG),正常饮食大鼠的FBG为(4.41±0.47)mmol/L,而高脂喂养大鼠的FBG为(6.17±1.65)mmol/L,较正常饮食组略有增加但无显著差异,见图2。

Figure 2. The levels of fasting blood glucose(FBG) in rats fed with high-fat diet(HF).±sE.n=6.
3高脂喂养大鼠空腹胰岛素浓度变化
采用放射免疫法测定大鼠空腹血清中胰岛素的浓度(fasting serum insulin, FSI)。高脂喂养12周后,大鼠空腹血清胰岛素浓度增加。结果见图3,正常饮食大鼠的FSI为(8.34±1.98) mU/L,而高脂喂养大鼠的FSI为(9.72±2.89) mU/L,较正常饮食组增加但无显著差异。
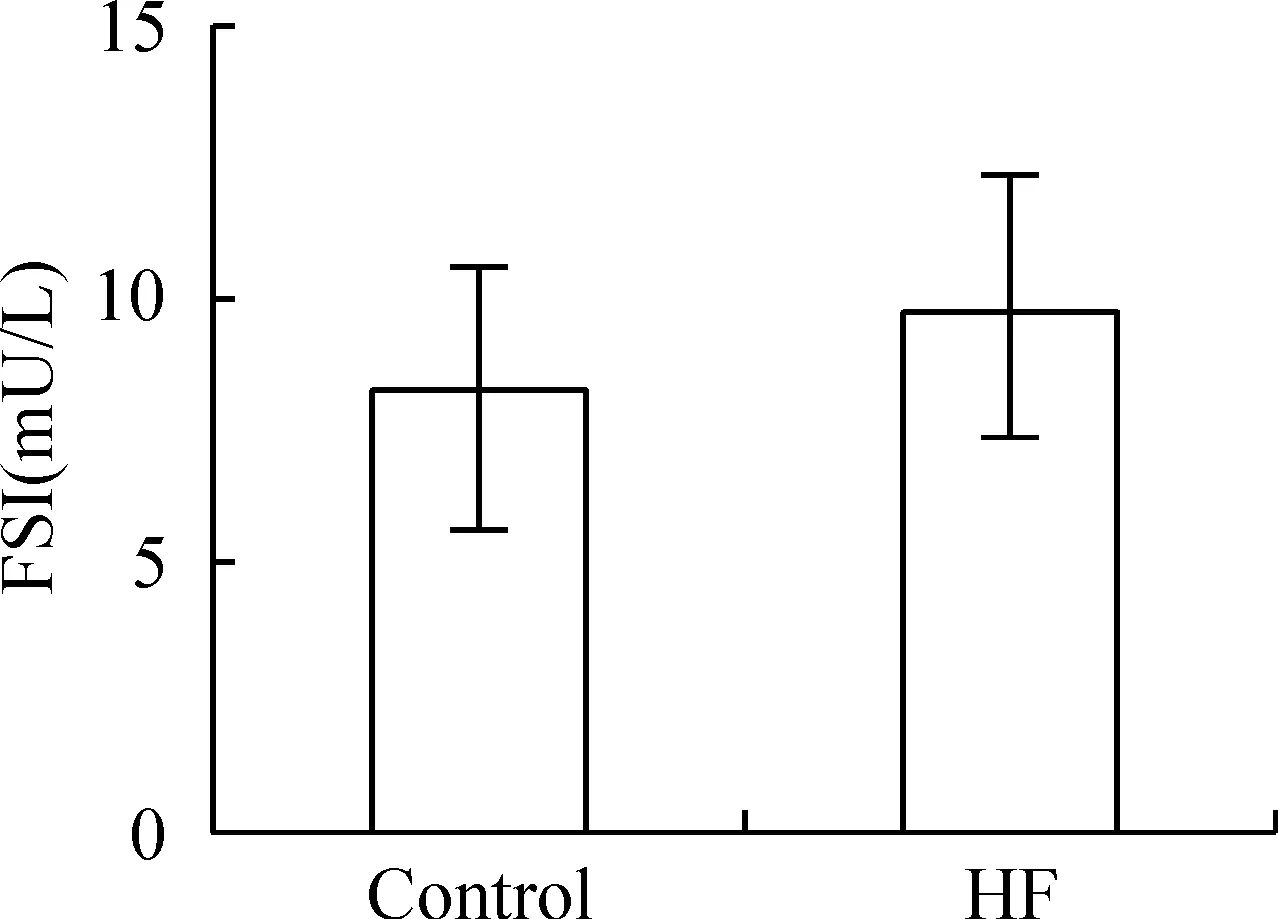
Figure 3. The levels of fasting serum insulin(FSI) in rats fed with high-fat diet(HF). ±sE. n=6.
4高脂喂养大鼠胰岛素敏感指数降低
根据空腹血糖浓度和血清胰岛素浓度,按照公式ln[1/(空腹血糖浓度×血清胰岛素浓度)]计算胰岛素敏感指数(insulin sensitivity index,ISI)。结果如图4所示,正常饮食大鼠的ISI为-3.85±0.57,而高脂喂养大鼠的ISI为-5.35±0.68,较正常饮食组显著降低(P<0.05),提示高脂喂养后大鼠胰岛素敏感性降低,产生了胰岛素抵抗。
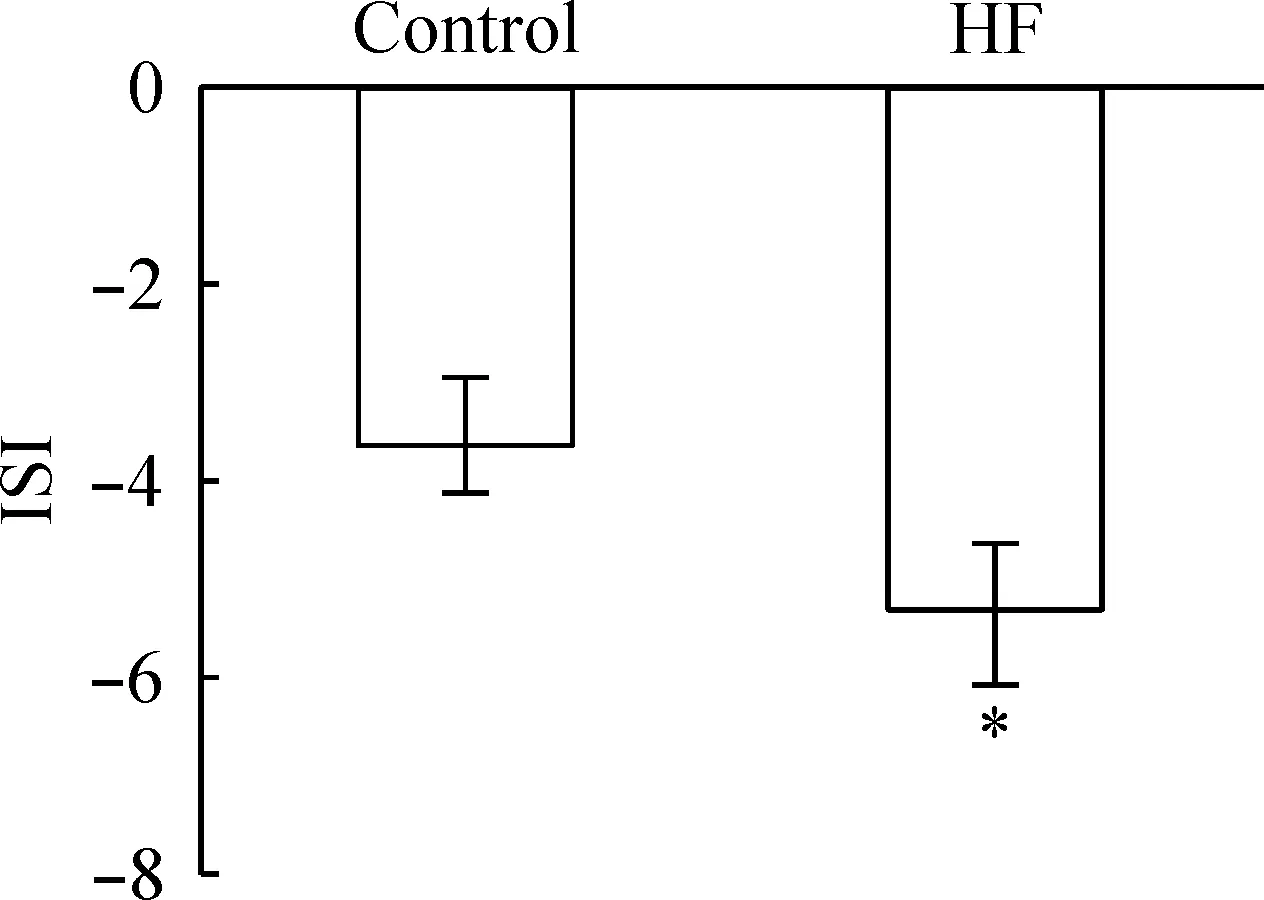
Figure 4. Decreased insulin sensitivity index(ISI) in rats fed with high-fat diet(HF). ±sE. n=6. *P<0.05 vs control.
5高脂喂养大鼠肝脏产生胰岛素抵抗
通过计算ISI,提示大鼠产生了胰岛素抵抗。而外周组织产生胰岛素抵抗的主要是肝脏、脂肪和肌肉。进一步观察大鼠肝脏组织产生胰岛素抵抗的情况。大鼠麻醉后处死,取新鲜肝脏组织,研磨后测定肝脏中的糖原含量。糖原合成降低是肝脏产生胰岛素抵抗的重要标志。高脂喂养12周后大鼠肝脏组织中糖原的含量为(13.75±2.22)mg/g,较正常饲料喂养对照组(23.60±3.76)mg/g显著降低(P<0.05),提示高脂喂养后大鼠肝脏组织产生了胰岛素抵抗,见图5。

Figure 5. Reduced glycogen content in liver tissues of rats fed with high-fat diet(HF). ±sE. n=6. *P<0.05 vs control.
6高脂喂养大鼠肝脏的活性氧产生变化
将大鼠用戊巴比妥钠麻醉后手术取肝脏组织标本OCT包埋后快速分别放入液氮中冷冻,冰冻连续切片,DHE(1∶1 000稀释)染色,免疫荧光显微镜下观察(1/100 s曝光时间)肝脏组织的ROS水平。结果见图6,高脂喂养大鼠肝脏组织中的ROS水平较正常对照组显著增加。
7高脂喂养大鼠肝脏的NOX3表达水平
用Western blotting分析肝脏组织的NOX3表达水平。结果显示高脂喂养大鼠肝脏组织中NOX3的表达显著升高,见图7。

Figure 6. Elevated ROS generation in liver tissues of rats fed with high-fat diet(HF)(×200).
Figure 7. Increased expression level of NOX3 in liver tissues of rats fed with high-fat diet(HF).±sE. n=3.*P<0.05 vs control.
讨 论
氧化应激在胰岛素抵抗和糖尿病发病中起重要作用。氧化应激是指体内ROS生成增加和(或)清除能力降低,导致ROS生成和清除失衡。 ROS具有重要的生理学作用,但过量的ROS会引起分子、细胞和机体的损伤。胰岛素抵抗是指胰岛素敏感的外周组织及靶器官或靶组织,主要是肝脏、脂肪组织、骨骼肌对胰岛素的敏感性及反应性降低,导致正常量的胰岛素产生的生物学效应低于正常水平。胰岛素抵抗是2型糖尿病发病的主要环节,其发生机制复杂,虽然进行了大量研究,但至今其分子机制尚未完全阐明。氧化应激在胰岛素抵抗中的作用机制倍受关注。大量研究都表明在各种胰岛素抵抗的动物和细胞模型中氧化应激水平上升,并且氧化应激激活的信号通路也导致胰岛素抵抗的发生和发展[3-5]。胰岛素抵抗主要发生在脂肪、肝脏和骨骼肌[6]。
在胰岛素抵抗的发生发展过程中,产生氧化应激的机制众多,主要有:1)糖自氧化:葡萄糖自身氧化作用增加,产生大量的ROS;2)蛋白质的非酶糖化:在非酶促条件下,葡萄糖和蛋白质相互作用形成 Amadori 产物,然后再形成晚期糖基化终末产物(advanced glycation end products, AGEs),AGEs通过与其受体RAGEs结合,促进ROS形成;3)多元醇通路的活性增高:醛糖还原酶多元醇代谢途径的活化,可降低 NADPH/NADP+,增加NADH/NAD+比例,消耗还原型GSH,从而诱导ROS合成[7,8];4)蛋白激酶C(protein kinase C, PKC)的活化:激活PKC,进而活化细胞NOX,诱导ROS的合成以及随后的脂质过氧化。反过来ROS也活化PKC,从而使ROS的产生进一步增加;5)抗氧化系统清除能力减弱:SOD、CAT、GSH-Px等抗氧化酶活性降低,维生素 C、维生素 E、GSH等抗氧化剂水平下降,体内抗氧化系统遭到破坏,明显削弱了机体清除ROS的能力。由此可见胰岛素抵抗的发生、发展过程中ROS产生增多和抗氧化能力减弱二者并存,从而发生氧化应激[9,10]。
我们的实验发现,高脂喂养的大鼠胰岛素敏感性降低,产生明显的胰岛素抵抗。而同时,大鼠肝脏组织也产生了胰岛素抵抗,其特点就是肝糖原含量显著降低,而肝脏的氧化应激水平升高。提示高脂喂养后的大鼠产生了胰岛素抵抗,并且高水平的氧化应激,进一步加重肝脏胰岛素抵抗状态。目前的研究认为ROS产生的途径有多种,肝细胞内ROS产生的主要酶体包括:NOX、线粒体电子传递链酶复合体、去偶联的一氧化氮合酶、黄嘌呤氧化酶等。其中NOX是肝细胞产生ROS的主要来源,NOX家族的分布具有组织特异性,最早在吞噬型细胞(粒性白细胞、单核/巨噬细胞)中发现了NOX2,后来又在各种组织和器官中发现新的同工酶,分别命名为NOX1、NOX3、NOX4、NOX5、Duox1和Duox2,它们均包含6个跨膜结构域和血红素、NADPH、FAD结合位点。NOX是细胞内ROS产生的主要途径之一。NOX主要还包括5个亚组分: p22phox、p47phox、p67phox、Rac1以及gp91phox。p22phox和gp91phox为膜蛋白,在中性粒细胞分泌小泡或特异的颗粒膜表面形成异源二聚体, 即细胞色素b558。NADPH的电子就是通过它的2种辅基由黄素蛋白FAD经血红素转移到O2上的[11,12]。肝脏主要表达NOX3及其亚组分p22phox、p47phox、p67phox和Rac1。NOX3是肝脏细胞中主要的NADPH氧化酶[13]。高脂饮食后,大鼠肝脏组织中NOX3的表达显著增加,提示NOX被激活,从而产生大量ROS,促进肝脏组织的氧化应激。我们的研究结果表明NOX源性的ROS在肝脏组织产生氧化应激和胰岛素抵抗中可能发挥了重要作用,但NOX源性ROS导致胰岛素抵抗的分子机制仍需要进一步深入探讨。
[1] Song F, Jia W, Yao Y, et al. Oxidative stress, antioxidant status and DNA damage in patients with impaired glucose regulation and newly diagnosed Type 2 diabetes[J]. Clin Sci(Lond), 2007,112(12):599-606.
[2] Kaneto H, Katakami N, Kawamori D, et al. Involvement of oxidative stress in the pathogenesis of diabetes[J]. Antioxid Redox Signal, 2007,9(3):355-366.
[3] Chang T, Wang R, Wu L, et al. Methylglyoxal-induced nitric oxide and peroxynitrite production in vascular smooth muscle cells[J]. Free Radic Biol Med, 2005, 38(2): 286-293.
[4] Chakraborti T, Das S, Mondal M, et al. Oxidant, mitochondria and calcium: an overview[J]. Cell Signal,1999,11(2):77-85.
[5] Leiter LA, Lewanczuk RZ. Of the renin-angiotensin system and reactive oxygen species Type 2 diabetes and angiotensin II inhibition[J]. Am J Hypertens, 2005,18(1): 121-128.
[6] 郭江红,韩德五,郭建红,等.内毒素诱发大鼠非酒精性脂肪性肝炎和胰岛素抵抗[J].中国病理生理杂志,2010,26(5):1009-1011.
[7] Dunlop M. Aldose reductase and the role of the polyol pathway in diabetic nephropathy[J]. Kidney Int Suppl, 2000,77(9):S3-S12.
[8] Inoguchi T, Li P, Umeda F, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells[J]. Diabetes, 2000,49(11):1939-1945.
[9] Masato K. Insulin resistance and atherosclerosis[J]. J Clin Invest, 2006,116(7):1756-1760.
[10]Semenkovich CF. Insulin resistance and atherosclerosis[J]. J Clin Invest, 2006,116(7):1813-1822.
[11]Babior BM. NADPH oxidase: an update[J]. Blood, 1999, 93(5): 1464-1476.
[12]Bissonnette SA, Glazier CM, Stewart MQ, et al. Phosphatidylinositol 3-phosphate-dependent and -independent functions of p40phoxin activation of the neutrophil NADPH oxidase[J]. J Biol Chem, 2008, 283(4): 2108-2119.
[13]Li L, He Q, Huang X, et al. NOX3-derived reactive oxygen species promote TNF-α-induced reductions in hepatocyte glycogen levels via a JNK pathway[J]. FEBS Lett, 2010,584(5):995-1000.
Mechanismofhepaticinsulinresistanceinducedbyhigh-fatdiet
LI Lan-fang, GUO Yu, TANG Guo-tao, CAO Xuan, YU Cui-yun, CHEN Lin-xi
(InstituteofPharmacyandPharmacology,NanhuaUniversity,Hengyang421001,China.E-mail:chenlinxi@tom.com)
AIM: To observed the relationship between oxidative stress and development of insulin resistance in hepatic tissues of Sprague dawley(SD) rats by analyzing reactive oxygen species(ROS) level and NADPH oxidase 3(NOX3) expression in livers.METHODSFour-week-old male SD rats were fed with high-fat diet containing 20% fat and 20% sucrose for 12 weeks to induce insulin resistance. Plasma insulin level was detected by radioimmunoassay. The content of liver intracellular glycogen was measured using a glycogen assay kit. ROS generation in the liver tissues was assessed by dihydroethidium(DHE) fluorescence. The expression of NOX3 was determined by Western blotting.RESULTSAfter 12 weeks of high-fat diet feeding, the content of blood glucose was increased but still maintained in normal level in the rats. However, the index of insulin sensitivity obviously decreased. Hepatic glycogen content in the rats fed with high-fat diet was significantly decreased, indicating that insulin resistance developed. Enhanced ROS production in hepatic tissues of the rats fed with high-fat diet was observed. Importantly, the expression of NOX3 in the liver was up-regulated in response to high-fat dietinvivo.CONCLUSIONHigh-fat diet feeding decreases insulin sensitivity, enhances ROS level and NOX3 expression, and reduces glycogen content in the livers.
Insulin resistance; NADPH oxidase; Oxidative stress
R363
A
10.3969/j.issn.1000-4718.2011.02-019
1000-4718(2011)02-0310-05
2010-08-05
2010-11-16
国家自然科学基金资助项目(No.30901577);湖南省衡阳市科技局资助项目(No.2009KJ14)
△通讯作者 Tel:0734-8281408;E-mail:chenlinxi@tom.com
猜你喜欢
广西糖业(2022年5期)2022-11-24
中老年保健(2021年5期)2021-08-24
小雪花·成长指南(2021年2期)2021-05-20
中华养生保健(2020年5期)2020-11-16
天津医科大学学报(2019年3期)2019-08-13
初中生世界·九年级(2019年4期)2019-05-05
实用肿瘤学杂志(2019年5期)2019-02-10
成都体育学院学报(2017年1期)2017-02-21
中国运动医学杂志(2016年3期)2016-07-10
医学研究杂志(2015年12期)2015-06-10
