
p型透明导电材料BaSnO3的第一性原理研究*
2011-11-02 03:26:37谭兴毅陈长乐金克新
物理学报 2011年10期
谭兴毅陈长乐金克新
p型透明导电材料BaSnO3的第一性原理研究*
谭兴毅 陈长乐金克新
(西北工业大学凝聚态结构与性质陕西省重点实验室,西安710129)
(2010年12月24日收到;2011年2月15日收到修改稿)
基于密度泛函理论,从头计算了N以及N和Sb共掺BaSnO3的电子结构和光学性质.结果表明N单掺BaSnO3与N和Sb共掺BaSnO3均为p型透明导电材料,在可见光区透过率均在80%以上,且N和Sb共掺具有更高的电导率.计算结果为实验上制备p型钙钛矿结构透明导电材料提供了强有力的理论指导.
p型透明导电材料,电子结构,光学性质
PACS:71.20.Ps,71.15.Mb,78.20.Ci
1.引言
钙钛矿结构氧化物由于具有简单的晶体结构和丰富的物理特性,如高温超导电性,巨磁电阻性,铁电性,多铁性,磁电效应等,成为目前研究最广泛的功能材料[1—4].在利用这些新奇物理特性制备薄膜器件过程中,不可避免的会用到电极材料.然而,目前所发现的大部分透明导电氧化物材料(TCO),如SnO2,In2O3,ZnO等[5—8],晶格结构都比较复杂,而且与钙钛矿结构氧化物具有较大的晶格失配,这种失配不利于薄膜之间相互生长.因此,寻找具有简单立方钙钛矿结构的TCO材料,成为制备全钙钛矿光电薄膜器件的必然趋势.目前文献所报道的钙钛矿型TCO材料有掺La,Sb,In的SrTiO[9—12]3,掺Nd的CaTiO[13]3,Cd3TeO[14]6等.BaSn O3是一种典型的立方钙钛矿结构氧化物,为n型宽带隙半导体材料,其禁带带隙为3.4 eV,并在1000℃依然具有很好的热稳定性[15],其晶格参数为a=4.123[16].但是未掺杂的BaSnO3的电阻率在103Ωcm以上,基本可认为是绝缘材料[15].近年来,以Parkash和Upadhyay等对BaSnO3块材和薄膜进行了A位的La,Sr掺杂和B位的Nb,Sb,Fe,Co,Ni,Cr等掺杂[17—20],从而得到了良好的导电特性,极大地丰富其在工业和科研中的应用.以上报道均为n型透明导电材料;但在薄膜器件中p型透明导电材料同样必不可少,所以寻找具有优良的p型透明导电材料成为制备薄膜器件的关键.本文利用第一性原理计算了N以及N,Sb共掺的BaSnO3的电子结构,发现N以及N,Sb共掺的BaSnO3具有p型导电特征,且N,Sb共掺的BaSnO3具有较大的电导率.
2.计算方法与模型
BaSnO3的晶胞常数a=0.4123 nm,是简单立方结构[16],几何结构优化后的晶格常数为0.4061 nm,实验和理论计算的晶格参数非常接近,误差为1.5%,表明该计算方法可行,所得结果合理.本文计算是在优化后的BaSnO3原胞基础上,在a,b和c基矢方向上分别扩展一个单位得到的2×2×2超晶胞,共含8个BaSn O3.使用替代法掺杂,对于N单掺杂的BaSnO3超晶胞模型,用一个N原子替代一个O原子,如图1(a)所示(N原子用圆圈勾画出);对于N和Sb共掺的BaSn O3超晶胞模型,用两个N原子和一个Sb原子分别替代两个O原子和一个Sn原子,具体替代位置如图1(b)所示(N原子用圆圈勾画出,Sb原子用矩形勾画出).计算工作采用基于密度泛函理论(density functional theory,DFT)结合平面波赝势方法的CASTEP (cambridge serial total energy paekage)软件[21]完成的.计算中采用周期性边界条件,用局域密度近似(LDA/CA-PZ)处理交换关联泛函,超软赝势处理离子实与价电子之间的相互作用,平面波基组描述体系电子的波函数.在计算时,先进行模型的几何结构优化,在最优几何结构下再进行单点能和电子结构计算.所有计算均在晶体倒易空间进行,最大截止能量为400 eV,计算收敛精度控制在1× 10-5eV/atom.布里渊区K矢量的选取为6×6× 6,晶体内应力收敛标准为0.05 GPa,原子平均受力不大于0.001 eV/nm.

图1 (a)N单掺杂的BaSnO3超晶胞模型;(b)N和Sb共掺的BaSnO3超晶胞模型
3.计算结果与讨论
3.1.掺杂体系的形成能
首先计算了结构优化后N掺杂以及N和Sb共掺BaSnO3超晶胞的形成能,其定义分别为

其中E(Ba8Sn8O23N)和E(Ba8Sn8O22N2Sb)为掺杂超晶胞的总能量,E(Ba8Sn8O24)为未掺杂超晶胞的总能量,E(N),E(O),E(Sb),E(Sn)为单个孤立原子的化学势.EN(form)和ENSb(form)能量值分别5.38和9.99 eV,说明N掺杂以及N和Sb共掺的BaSnO3均可以在实验中制备.
3.2.电子结构
图2(a),(b),(c)和图3分别为纯BaSn O3,N掺杂BaSnO3和N和Sb共掺BaSnO3体系的能带图,电子总态密度以及部分原子的分波态密度图,图中零点为费米能级.由于远离费米面的电子不影响导电性,故本文能带图和电子态密度图均只给出了能量在-5与8 eV之间的部分.从图2(a)看出,纯BaSnO3是一种间接禁带半导体材料(R—G),与实验相符,其禁带宽度为0.955 eV,小于实验值3.4 eV[15].原因在于计算中采用的DFT是基态理论,而能隙属于激发态,因此得到的结果偏低,这也是采用该理论计算时普遍存在的现象,但这并不影响对BaSnO3电子结构的理论分析.在纯BaSnO3中图2 (a),图3(a)邻近费米面的价带主要来源于O-2 p电子态的贡献,导带主要由Sn的电子态组成,即Sn-O八面体结构决定着BaSnO3体系的导电性能.与纯BaSnO3的能带结构相比,N掺杂BaSn O3和N和Sb共掺BaSnO3体系的简并能级均发生分裂(如图2中的F点和Z点所示).从图3(b),(c)看出掺杂后体系总电子态密度向低能方向移动,电子态在布里渊区的F点和Z点发生了退简并化,而掺杂的N-2 p电子出现在费米面附近,使费米能级进入价带顶.对比图2(a),(b),(c),可以发现,禁带宽度从0.955 eV,0.498 eV减小到0 eV,即导电性随着掺杂显著增强,特别是N和Sb共掺BaSnO3体系比N掺杂BaSnO3体系的导电性有极大提高.从图3(b)可以发现由于N掺杂使得Sn-O八面体结构发生变化,Sn电子态向低能方向移动,降低了禁带宽度,增加了材料的导电性;从图3(c)看出Sn电子态同样向低能方向移动,并且N-2 p态电子和掺杂的Sb电子(Sb-5s,Sb-5 p态电子)产生杂化,从而使体系的禁带宽度更小,即杂质和缺陷带之间的重叠使带隙变窄[22].杂质原子的电子对掺杂体系起着极大的作用.对于费米能级附近的态密度主要来源于N-2 p,Sb-5p态电子的贡献,明显改善了BaSnO3的导电性能,因此我们认为N和Sb是实现低阻BaSnO3材料的合适掺杂元素.依据掺杂理论,当掺杂高于一定浓度时,原来位于禁带中的尖锐的分立施主能级和受主能级都可以扩展成杂质能带并与导带有所交叠,使导电性极大的提高.
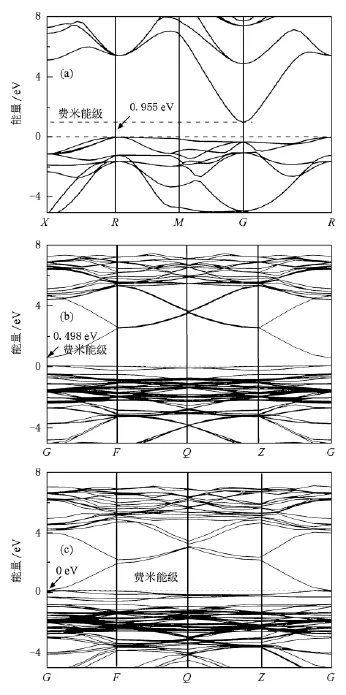
图2 体系的能带图(a)纯BaSnO3;(b)N掺杂BaSnO3;(c) N和Sb共掺BaSnO3
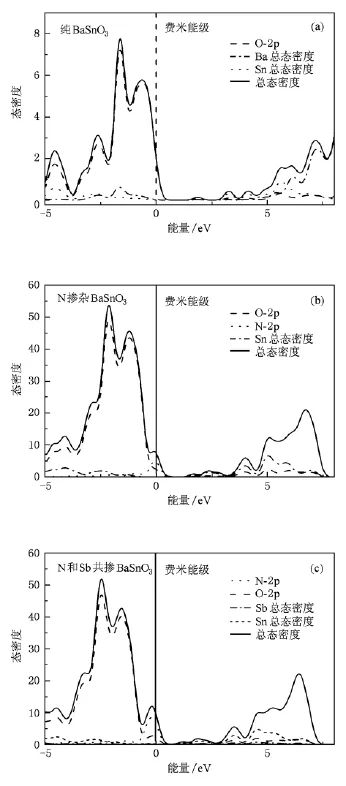
图3 电子总态密度以及部分原子的分波态密度图(a)纯BaSnO3;(b)N掺杂BaSnO3;(c)N和Sb共掺BaSnO3
3.3.光学特性
体系在较小波矢下对光场的响应为线性,在此响应范围内固体宏观光学响应函数可用光的复介电常数ε(ω)=ε1(ω)+iε2(ω)或者复折射率N(ω)=n(ω)+i K(ω)描述,其中ε1=n2-K2,ε2=2 n K.由于计算电子结构中无论是带间还是带内跃迁频率都远超过声子频率,而且使用的方法是单电子近似法,故仅考虑电子激发.从量子力学的观点看,带间跃迁光吸收过程是电子在辐射电磁场微扰作用下从低能态跃迁到高能态的过程.根据直接跃迁概率的定义和克拉默斯克勒尼希(Kramers-Kronig)色散关系可以推导出晶体介电函数虚部ε2(ω),实部ε1(ω),折射率n(ω),消光系数k(ω),反射系数R(ω),吸收率η(ω),透过率T(ω)等[23—25]
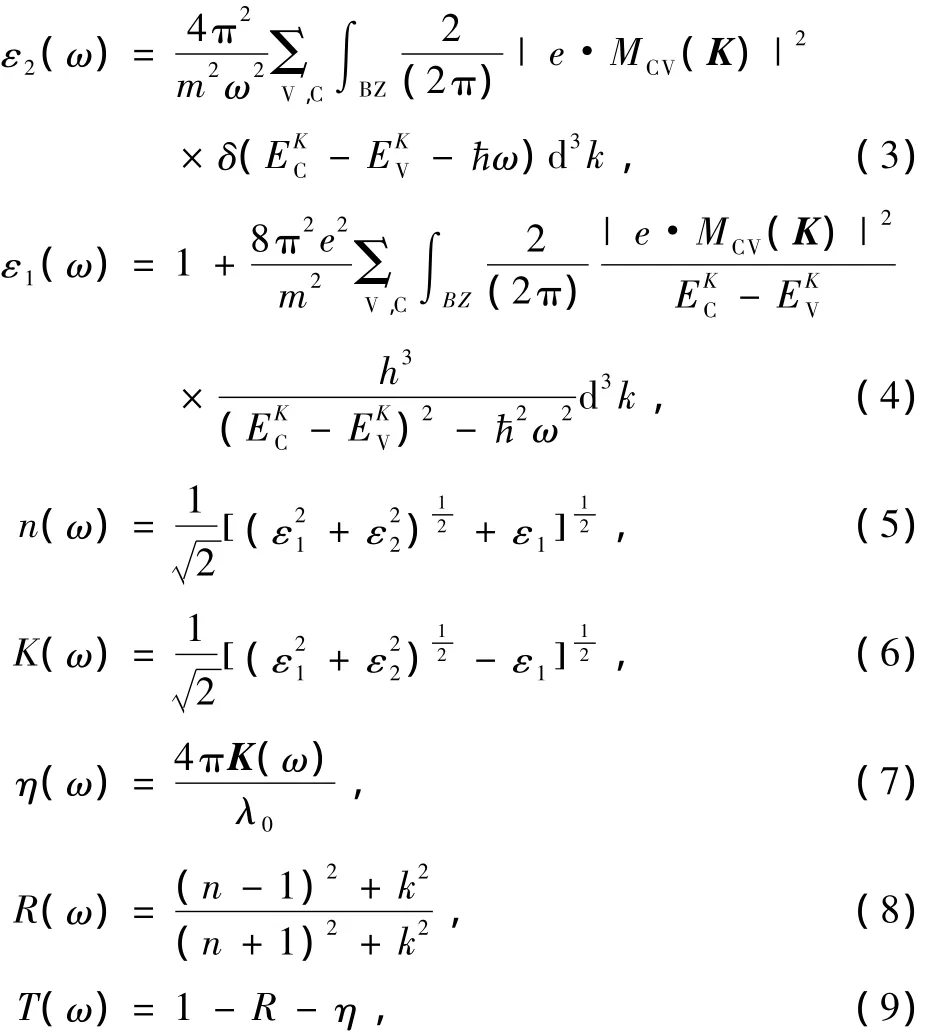
其中C,V分别表示导带和价带,BZ为第一布里渊区,K为倒格矢,为普朗克常数,|e·MCV(K)|2为动量跃迁矩阵元,ω为圆频率,λ0为光在真空中的波长,EKC和EKV分别为导带和价带的本征能级.以上关系是分析晶体能带结构和光学性质的理论依据,它反映了能级间电子跃迁所产生光谱的发光机理.从某种意义上说,复介电函数ε(ω)比宏观光学常数更能表征材料的物理特性,更易于与物理过程的微观模型及固体的微观电子结构联系起来.图4为纯BaSnO3,N掺杂BaSn O3和N和Sb共掺BaSnO3体系光学性质的计算结果.纯BaSn O3,N掺杂BaSnO3和N和Sb共掺BaSnO3体系在波长范围为300到800 nm的可见光区透过率均高于80%,并按照纯BaSnO3,N掺杂BaSnO3,N和Sb共掺BaSnO3的顺序透过率依次增加,反射率依次减小.三者之间吸收率按照纯BaSn O3,N掺杂BaSnO3,N和Sb共掺BaSn O3的顺序依次增加,源于杂质原子N和Sb在费米能级处态密度较O原子以及Sn原子高,使得电子跃迁概率率增加,光吸收增强,计算结果如表1所示.但由于吸收率数值远小于反射率(相差一个数量级),所以材料光学透过率的增加主要源于其反射率的减小.
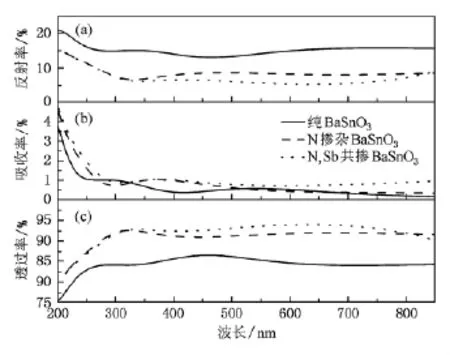
图4 纯BaSnO3,N掺杂BaSn O3和N和Sb共掺BaSnO3体系反射率,吸收率和透过率的计算结果
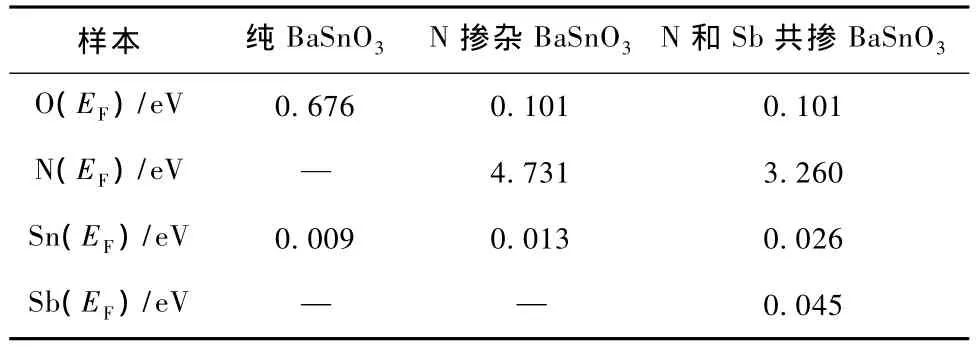
表1 费米能级处态密度值
4.结论
本文利用第一性原理从头计算了N以及N和Sb共掺BaSnO3的形成能,能带结构,态密度分布以及光学性质.计算结果表明N掺杂以及N和Sb共掺的BaSnO3可在实验中制备,掺杂明显改善了BaSnO3体系的导电性能.计算结果给p型BaSnO3透明导电材料的制备提供了理论基础,具有极为重要的指导意义.
[1]TokuraY 2006 Science 312 1481
[2]Ramesh R,Spaldin A N 2007 Nature Mater 6 21
[3]Jin S,Tiefel T H,McCormack M,Fastnacht R A,Ramesh R Chen L H 1994 Science 264 413
[4]Sacchetti A,Guidi M C,Arcangeletti E,Nucara A,Calvani P,Piccinini M,Marcelli A,Postorino P 2006 Phys.Rev.Lett.96 35503
[5]HoSono H 2007 Thin Solid Films 515 6000
[6]Minami T 2000 MRS Bull 25 38
[7]Bernardi M L,Soledade L E,Santos I A 2002 Thin Solid Films 405 228
[8]Park Y,Choong V,Gao Y 1996 Appl.Phys.Lett.68 2699
[9]Cho J H,Cho H J 2001 Appl.Phys.Lett.79 1426
[10]Wang H H,Chen F,Dai S Y 2001 Appl.Phys.Lett.78 1676
[11]Guo H Z,Liu L F,Fei Y Y 2003 J.Appl.Phys.94 4558
[12]Wang H H,Cui D F,Dai S Y 2001 J.Appl.Phys.90 4664
[13]Wang R P,Tao C J 2002 J.Crystal.Growth.245 63
[14]Tetsika H,Shan Y J,Tezuka K 2006 J.Vac.Sci.Technol.A 24 L4
[15]Ostrick B,Fleischer M,Meixner H 1997 J.Am.Ceram.Soc. 80 2153
[16]Hinatsu Y 1996 J.Solid State Chem.122 384
[17]Upadhyay S 2008 Phys.Stat.Sol.(a)205 1113
[18]Upadhyay S,Parkash O,Kumar D 2007 J.Electroceram 18 45
[19]Upadhyay S,Parkash O,Kumar D 2004 J.Phys.D:Appl.Phys.37 1483
[20]Wang H F,Liu Q Z,Chen F,Gao G Y,Wu W B,Chen X H 2007 J.Appl.Phys.101 106105
[21]Segall M D,Philip J D Lindan,M J Probert,Pickard C J,Hasnip P J,Clark S J,Payne M C.2002 J.Phys:Condens.Matter.14 2717
[22]Mahan G D,1980 J.Appl.Phys.51 2634
[23]Guo J Y,Zhang G,He K H,Chen J Z 2008 Acta Phys.Sin.57 3740(in Chinese)[郭建云、郑广、何开华、陈敬中2008物理学报57 3740]
[24]Xing H Y,Fan G H,Zhang Y,Zhao D G 2009 Acta Phys.Sin.58 0450(in Chinese)[刑海英、范广涵、章勇、赵德刚2009物理学报58 0450]
[25]Peng L P,Xu L,Yin J W 2007 Acta Phys.Sin.56 1585(in Chinese)[彭丽萍、徐凌、尹建武2007物理学报56 1585]
PACS:71.20.Ps,71.15.Mb,78.20.Ci
*Project supported by the National Natural Science Foundation of China(Nos.61078057,50702046),the NPU Foundation for Fundamental Research(Grant Nos.NPU-FFR-JC201048,JC200821),and NWPU“Aoxiang Star”Project.
Corresponding author.E-mail:chenchl@nwpu.edu.cn
p-type transparent conductive BaSnO3: A first-principles calculations*
Tan Xing-Yi Chen Chang-LeJin Ke-Xin
(Shannxi Key Laboratory of Condensed Matter Structures and Properties,School of Science,Northwestern Polytechnical University,Xi'an 710129,China)
(Received 24 December 2010;revised manuscript received 15 February 2011)
Based on density functional theory calculations,the electronic properties of N-doped BaSnO3and N and Sb codoping are investigated.It is found that codoping with N acceptors and Nb donors in a ratio of 2∶1 is suitable for the fabrication of low-resistivity p-type BaSnO3.Our results indicate that codoping with N acceptors and Nb donors is a prospective candidate as a p-type transparent conductive material.
p-type transparent conductive material,electronic structures,optical properties
*国家自然科学基金(批准号:61078057,50702046)、西北工业大学基础研究基金(批准号:NPU-FFR-JC200821,JC201048)和西北工业大学“翱翔之星”项目资助的课题.
.E-mail:chenchl@nwpu.edu.cn
猜你喜欢
高中数理化(2024年4期)2024-03-16 11:09:37
杭州(2023年3期)2023-04-03 07:22:04
高中数理化(2022年16期)2022-09-14 13:57:04
复旦学报(医学版)(2020年3期)2020-06-18 07:36:52
原子与分子物理学报(2020年5期)2020-03-17 07:00:00
中学生数理化(高中版.高考理化)(2019年10期)2019-11-08 03:23:36
物理学进展(2017年1期)2017-02-23 01:35:44
云南师范大学学报(自然科学版)(2015年5期)2015-12-26 12:46:14
太阳能(2015年4期)2015-02-28 17:08:19
太阳能(2015年2期)2015-02-28 17:07:18
