
聚合型硼氢化物(BH3)n(n=1—3)的几何结构与光谱的研究*
2011-11-02 03:26:38伍冬兰谢安东万慧军阮文
物理学报 2011年10期
伍冬兰谢安东万慧军阮文
聚合型硼氢化物(BH3)n(n=1—3)的几何结构与光谱的研究*
伍冬兰谢安东 万慧军 阮文
(井冈山大学数理学院,吉安343009)
(2010年11月1日收到;2010年12月7日收到修改稿)
采用不同方法B3P86,B3LYP,MP2和LSDA,结合Dunning的相关一致基组cc-PVTZ,对聚合型硼氢化物(BH3)n(n=1—3)分子的可能几何构型进行优化计算,得出最稳定构型的几何参数、电子结构、振动频率和光谱等性质参数,并给出了最稳定结构的总能量(ET),结合能(EBT),平均结合能(Eav),电离势(EIP),能隙(Eg),费米能级(EF)等.结果表明:采用密度泛函DFT中的方法B3P86计算的能量最低,结构参数更接近文献值;三种硼氢化物分子基态都为1重态,电子态分别为1A',1A和1A;BH3分子的最稳定几何构型为平面三角形结构;B2H6为对称性乙烯式D2h立体结构,H—B之间生成氢桥式三中心双电子键;B3H9为C3ν立体结构,也生成氢桥式三中心双电子键,但三个氢桥三中心双电子键彼此隔离.最后分析了三种氢化物的红外和拉曼光谱、平均结合能、电离势、能隙和费米能级等特性,说明(BH3)n(n=1—3)三分子中B2H6最稳定,H—B桥键键长比端键更长,最强峰红外光谱强度最大.
聚合型硼氢化物,几何构型,光谱
PACS:31.15.A-,33.15.Dj,33.15.Fm,36.40.Mr,36.40.Qv
1.引言
聚合型氢化物也称为多中心键氢化物,像Be,Mg,B,Al等元素的原子属于缺电子原子,他们能与等电子原子H化合,生成缺电子分子的化合物,即通过氢桥键形成含多中心键的氢化物,例如(BeH2)n,(BH3)n,(AlH3)n等[1].与其他氢化物相比,聚合型氢化物与水反应作用时会放出氢气,具有更低的形成能和更高的H/M(M=B,Al),因而具有更好的放氢热力学性质和更高的储氢量.这类氢化物与其他碱金属或碱土金属的离子型氢化物结合成的配位氢化物具有最高的储氢质量密度,是近年来研究最活跃的储氢材料[2—5].对于聚合型硼氢化物,BH3分子因为不能稳定存在研究不多;B2H6在373 K温度下稳定,高于此温度则分解放出H2,文献中对B2H6的几何构型做了细致的分析[6—9],而对其电子特性和光谱等研究涉及不多;B3H9分子结构中的氢桥式三中心双电子键由于彼此隔离[10],热稳性差,其电子特性和光谱的相关研究几乎未见文献报道.
本文采用Gaussian03程序包,采用不同方法B3 P86,B3 LYP,MP2和LSDA结合cc-PVTZ基组,对聚合型硼氢化物(BH3)n(n=1—3)可能的几何构型和多重性进行优化计算,得出最稳定构型的几何结构和相关的物理性质参数.结果表明,采用密度泛函B3P86方法计算的能量最低、结构更接近文献值;(BH3)n(n=1—3)分子属于缺电子原子B和等电子原子H化合,生成缺电子分子的氢化物,其中BH3分子的最稳定几何构型为平面三角形结构,但是该分子因为H原子没有电子对,不能与B原子的空P轨道形成P—Pπ键而不能稳定存在;B2H6为对称性乙烯式D2h立体结构,分子中的硼原子采用不等性的sp3轨道与另一硼原子的不等性sp3轨道及氢原子的1 s轨道交互重叠生成氢桥式三中心双电子键[6];B3H9为C3ν立体结构,硼原子与硼原子及氢原子的轨道交互重叠仍然是生成氢桥式三中心双电子键,但三个氢桥三中心键由于彼此隔离,没有形成更大的多中心键的条件,仍保持为三个三中心双电子键[10].最后分析了三种氢化物的红外和拉曼光谱、自然电荷分布、平均结合能、电离势费米能级等特性,说明聚合型氢化物(BH3)n(n=1—3)中B2H6最稳定,H—B桥键比端键更长,最强峰红外光谱强度最大.
2.理论计算方法
本文在Gaussian03程序基础上,采用不同方法B3P86,B3 LYP,MP2和LSDA,在Dunning的相关一致基组cc-PVTZ水平上分别对BH3,B2H6和B3H9三分子各种可能的几何结构、多重性为1,3,5进行优化计算,得出采用密度泛函方法B3P86的结果更接近文献值、能量也最低.再利用B3P86方法对他们的+1价离子的各种可能的几何结构、多重性为2,4多种情况进行几何优化和频率计算,根据能量最低原理,找出各分子的能量最低对应的几何构型,即基态构型.对正一价离子进行单点能扫描,平衡几何参数从正一价分子离子的基态平衡几何参数到中性分子平衡几何参数扫描,用正一价离子在中性分子平衡几何参数时的能量与基态中性分子平衡几何参数的能量之差为第一垂直电离能(EIP);所有中性原子的能量之和与中性分子的总能量之差为结合能(EBT)[11];结合能与原子数n的比值为平均结合能(Eav);最高占据轨道能量与最低空轨道能量之差为能隙(Eg);最高占据轨道的能量为费米能级(EF)[12].图1为(BH3)n(n=1—3)已优化的基态稳定结构,图2和图3分别为本文计算的红外、拉曼光谱和电子特性.表1和表2分别列出了(BH3)n(n=1—3)的几何结构参数和自然电荷布居分布.
3.计算结构与讨论
3.1 .平衡几何结构
采用不同方法B3 P86,B3LYP,MP2和LSDA,结合Dunning相关一致基组cc-PVTZ,对(BH3)n(n= 1—3)所有可能的几何结构和多重性进行键长和键角全面优化计算.根据能量最低原理得到了(BH3)n(n=1—3)最稳定基态结构,如图1所示,括号内为所属点群和结合能.从图1可知,BH3是平面结构,属于Cs群,其电子态为1A';B2H6为对称性乙烯式的立体结构,属于D2h群,其电子态为1A,其中连在B原子上的H形成共价键(端键),且都在同一平面上,而垂直于该平面的H原子则与B原子形成氢桥键(桥键),构成三中心双电子键;B3H9是立体结构,属于C3ν群,其电子态为1A,从图上可看出也形成了三个氢桥三中心双电子键,但彼此隔离,氢桥键之间作用较弱.表1中列出了采用不同方法B3 P86,B3 LYP,MP2和LSDA对(BH3)n(n=1—3)优化计算的最稳定结构的能量、键长和键角,其中括号内为文献值[6,13].从表中数据可看出,本文采用方法中,B3 P86方法优化计算的三个分子构型的能量最低,这与文献[6]的结果一致;而且与文献相比,本文计算的能量更低,键长和键角与文献接近,且桥键都比端键要长,说明我们采用B3 P86方法和cc-PVTZ基组进行计算比较可靠,优化计算出来的几何构型可以用来进一步分析(BH3)n(n=1-3)氢化物的光谱和电子特性.
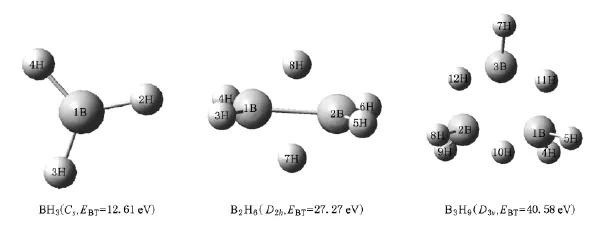
图1 (BH3)n(n=1—3)最稳定的基态结构、所属点群和结合能
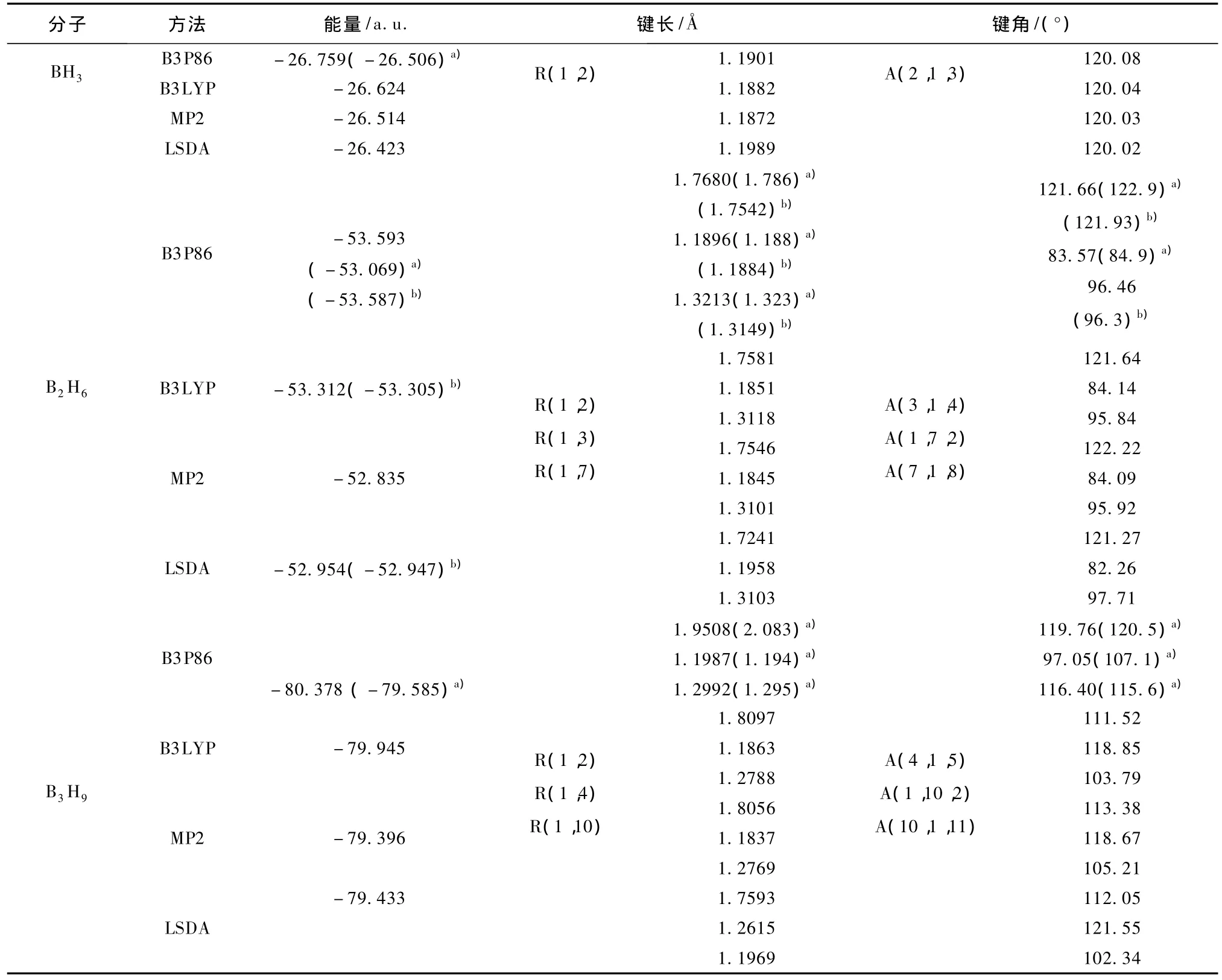
表1 (BH3)n(n=1—3)氢化物最稳定构型的几何参数
3.2 .振动光谱
利用B3 P86方法在cc-PVTZ水平上,对(BH3)n(n=1—3)氢化物最稳定结构的振动光谱进行了计算,其中红外IR和Raman光谱如图2所示.计算得到的振动频率均为正值,表明各硼氢化物分子最稳定结构均为势能面上的极小点.计算结果分析表明:BH3氢化物的IR光谱主要集中在1149.14—1195.27 cm-1和2695.79—2696.85 cm-1两段,其中最强振动峰在2695.79 cm-1,主要是H—B键的反对称伸缩振动,Raman光谱主要集中在2561.38—2696.85 cm-1段,其中最强振动峰在2561.38 cm-1,主要表现为H—B键的对称伸缩振动;B2H6氢化物的IR光谱主要集中在982.89—1187.16 cm-1,1700.06—1750.08 cm-1和2610.39—2715.27 cm-1三段,其中最强振动峰在1722.05 cm-1,主要表现为氢桥键上两个H原子对称平行于B—B所在平面的水平往复运动,这也说明了B2H6氢化物氢桥键的作用比较强烈,Raman光谱主要集中在815.29—1194.49 cm-1,1186.69—2191.33 cm-1和2623.85—2700.62 cm-1三段,其中最强振动峰在2523.85 cm-1,主要表现为H—B端键的对称伸缩振动;B3H9氢化物的IR光谱主要集中在1076.11—1163.51 cm-1,1854.22—1993.61 cm-1和2555.88—2725.24 cm-1三段,其中最强振动峰在2725.24 cm-1,主要表现为H—B端键的反对称伸缩振动,Raman光谱主要集中在1854.22—1993.61 cm-1和2555.88—2725.24 cm-1两段,其中最强振动峰在2565.93 cm-1,主要表现为H—B端键的对称伸缩振动,次强振动峰在1854.22 cm-1,主要表现为氢桥键的对称伸缩振动,这说明氢桥键的振动相对于端键的振动更弱,同时也说明该氢化物中的氢桥键因为彼此隔离作用减弱.从分析可看出,B2H6氢化物氢桥键的最强振动频率比B3H9大,这与前面优化得到的氢桥键键长前者大于后者的结论相符合.

图2 (a)为红外光谱IR(kM/mole);(b)为拉曼光谱Raman(4/AMU)
3.3.电荷
在B3 P86/cc-PVTZ水平上,采用自然键轨道(NBO)方法对(BH3)n(n=1—3)最稳定结构的自然电荷布居进行了分析,表2给出了(BH3)n(n= 1—3)各原子上的自然电荷分布.可以看出,对于B2H6和B3H9这两种配位氢化物,在原子相互作用形成氢化物的过程中,发生了较多的电荷转移,这种电荷转移的作用使得B原子呈负电性,H原子显正电性.B原子的自然电荷在-0.156—-0.324之间,其中B2H6中B原子的自然电荷为-0.158,文献中为-0.2[14,15],H原子的自然电荷在0.016—0.186之间,B—H之间呈现共价键特性,其中氢桥键上H原子的自然电荷比端键上的电荷强得多,这说明形成氢化物时氢桥键上H原子更稳定,不容易断裂.

表2 (BH3)n(n=1—3)最稳定构型中各原子的电荷
3.4 .稳定性
采用B3 P86方法在cc-PVTZ水平上,对(BH3)n(n=1—3)分子的EBT,EIP,Eg,Eav和EF进行了计算.图3中给出了(BH3)n(n=1—3)三种分子的EIP,Eg,Eav和EF变化关系.从图中可以看出,BH3分子具有相对较大的电离势,以及相对较低的费米能级,但由于H原子没有电子对,不能与B原子的空P轨道形成(P—P)π键而不能单独稳定存在,一般是两个BH3分子以氢桥键的形式结合形成B2H6分子存在;而对B2H6和B3H9两分子来说,B2H6的电离势和平均结合能都比B3H9大,说明B2H6分子相对来说更加稳定,主要是由于氢桥键的作用前者比后者强的原因,进一步说明B3H9分子中的氢桥键由于彼此隔离而作用变弱.
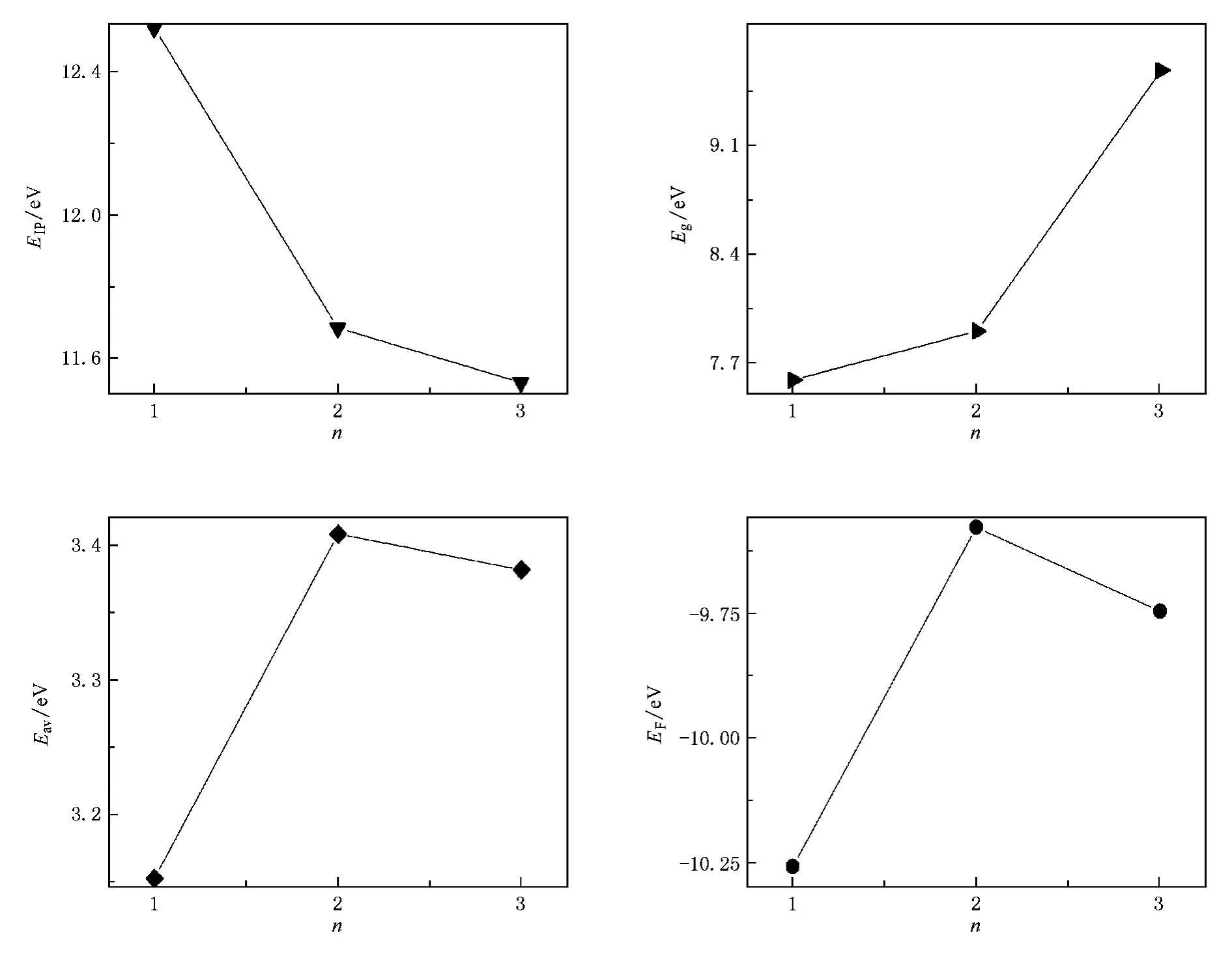
图3 (BH3)n(n=1—3)分子的EIP,Eg,Eav和EF
4.结论
采用不同方法优化计算了聚合型硼氢化物(BH3)n(n=1—3)分子可能的几何构型,得出最稳定构型的几何参数、电子结构、振动频率和光谱、电子特性等性质.计算结果表明,BH3分子为平面三角形结构,但由于H原子没有电子对,不能与B原子的空P轨道形成(P—P)π键而不能单独稳定存在; B2H6为对称性乙烯式D2h立体结构,H—B之间生成了氢桥式三中心双电子键,稳定性最高;B3H9为 C3ν立体结构,也生成氢桥式三中心双电子键,但三个氢桥三中心双电子键由于彼此隔离而热稳性差.最后分析了三种氢化物的红外和拉曼光谱、电荷分布、平均结合能、电离势和费米能级等的特性,进一步说明(BH3)n(n=1—3)化合物中B2H6最稳定,H—B桥键键长比端键更长,最强峰红外光谱强度最大.本文研究为进一步研究该类氢化物的储氢性能提供理论参考;对研究它们与其他碱金属或碱土金属的离子型氢化物结合成配位氢化物储氢材料也具有一定的参考价值.
[1]Liu Q L,Yue X X 2004 Journal of Xinxiang Teachers College 18 13(in Chinese)[刘清玲、岳小欣2004新乡师范高等专科学校学报18 13]
[2]Chen J,Zhu M 2009 Mater Ials China 28 2(in Chinese)[陈军、朱敏2009中国材料进展28 2]
[3]Zhang J,Bai C G,Pan F S,Luo X D 2008 Ordnance Material Science and Engineering 31 90(in Chinese)[张静、白晨光、潘复生、罗晓东2008兵器材料科学与工程31 90]
[4]Xiao X Z 2008 A D.Ph.Dissertation(Zhejiang University)(in Chinese)[肖学章2008博士学位论文(浙江大学)]
[5]Hou Y Q,Zhang X D,Jiang Z Y 2010 Aata Phys.Sin.59 5667 (in Chinese)[侯榆青、张小东、姜振益2 0 1 0物理学报5 95667]
[6]Yan S Y,Ma M Z,Zhu Z H 2005 Aata Phys.Sin.54 3106(in Chinese)[阎世英、马美仲、朱正和2005物理学报54 3106]
[7]Sidgwick N V 1927 The Electronic Theory of Valency(Oxford: Clarendon Press)
[8]Dilthey W Z 1921 Angew.Chem.34 596
[9]Lemi T 2003 Journal of Molecular Structure(Theochem)629 279
[10]Wu H S,Pan D K,Zhou W L,Liu Y L 1996 Acta Chim.Sin.54 638(in Chinese)[武海顺、潘道皑、周伟良、刘元隆1996化学学报54 638]
[11]Sheng X H,Zhu Z H,Gao T,Luo S Z 2006 Aata Phys.Sin.55 3420(in Chinese)[谌晓洪、朱正和、罗顺忠2006物理学报55 3420]
[12]Mao H P,Wang H Y,Ni Y,Xu G L,Ma M Z,Zhu Z H,Tang Y J 2004 Aata Phys.Sin.53 1766(in Chinese)[毛华平、王红艳、倪羽、徐国亮、马美仲、朱正和、唐永建2004物理学报53 1766]
[13]Brian J D,Liang C X,Henry F S III 1991 J.Am.Chem.Soc.113 2884
[14]Hamilton W C 1956 Proc.Roy.Soc.(London)A 235 359
[15]Yamazaki M 1957 Chem.Phys.27 1401
PACS:31.15.A-,33.15.Dj,33.15.Fm,36.40.Mr,36.40.Qv
*Project supported by the National Natural Science Foundation of China(Grand No.10965002),and the Scientific Research Program of the Education Bureau of Jiangxi Province,China(Grand Nos.2006263,2007326).
E-mail:wudonglan1216@sina.com
Study on geometrical structure and spectrum of polymerization borohydride(BH3)n(n=1—3)*
Wu Dong-LanXie An-Dong Wan Hui-Jun Ruan-Wen
(College of Mathematic and Physical,Jinggangshan University,Ji’an 343009,China) (Received 1 November 2010;revised manuscript received 7 December 2010)
The possible geometrical structures of polymerization borohydride(BH3)n(n=1—3)are optimized in computation,based on different methods of B3 P86、B3 LYP,MP2,LSDA and by combining the Dunning relevant and consistent base group cc-PVTZ.The configuration geometric parameter,the electronic structure,the vibrational frequency and spectrum of the most stable structure are obtained,and the total energy(ET),binding energy(EBT),the average binding energy (Eav),the ionization potential(EIP),the energy crack(Eg),the Fermi level(EF)and so on are also given.The results indicate that the total energy is lowest and its value is close to the reported values from B3 P86 method.The ground state of the three kinds of borohydride are all singlet states,the ir electronic states respectively are1A',1A,and1A.The stable geometry configuration of BH3molecule is the planar triangle,B2H6has a symmetrical ethylene type D2hspatial structure,and between H-B produces the hydrogen bridge type with three-center double electronic key,B3H9has a C3νspatial structure,also produces a hydrogen bridge type of three-center double electronic key,but the three hydrogen bridge types are isolated from each other.Finally the infrared and the Raman spectrum,the average binding energy,the ionization potential,the energy gap,Fermi level and so on are analyzed.B2H6is shown to be the most stable molecule in(BH3)n(n=1—3),the H-B bridge bond key long is longer than the terminal lond,the infrared intensity of strongest peak is a maximal value.
polymerization borohydride,geometrical structure,spectrum
*国家自然科学基金(批准号:10965002)和江西省教育厅科学技术项目(批准号:2006263,2007326)资助的课题.
E-mail:wudonglan1216@sina.com
猜你喜欢
山东冶金(2022年4期)2022-09-14 08:59:08
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 06:42:32
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08 00:48:08
经济技术协作信息(2018年11期)2019-01-14 03:07:22
环境保护与循环经济(2017年12期)2017-07-11 01:46:36
北京航空航天大学学报(2017年10期)2017-04-20 08:51:23
航天返回与遥感(2014年4期)2014-07-31 17:47:47
无机化学学报(2014年4期)2014-02-28 17:31:08
