
尖晶石型锂离子筛的制备及其吸附性能
2011-09-17 07:34:34
中南大学学报(自然科学版) 2011年8期
(中南大学 冶金科学与工程学院,湖南 长沙,410083)
锂是国民经济和国防建设中重要的战略资源[1]。锂在新型高能电池、航空航天技术、超轻高强度的锂铝合金和核聚变发电等领域有广泛的应用[2−3]。自然界的锂资源主要赋存于海水、盐湖卤水、地热水和花岗伟晶岩型矿床中,其中矿石中锂的储量不足总储量的3%。从长远意义看,陆上锂资源将不能满足日益增长的需求,因此,迫切需要开发盐湖卤水、海水和地热水等稀锂液态资源[4]。从稀锂溶液中提取锂以离子交换与吸附法最为绿色经济已成共识[5],该技术的关键就是制得吸附性能良好的锂离子筛(又称锂吸附剂)。国内外在锂离子筛的制备方面已经取得了一定的成绩,报道已合成的经处理后有吸附能力的氧化物有LiMn2O4,Li1.6Mn1.6O4[6],Li1.33Mn1.67O4[7],LiMnO[8]和 LiMnO2[9]等,这些锰氧化物在一定浓度的酸(如盐酸、硫酸、硝酸等)下洗涤,洗去锂离子,便可得到锰系离子筛,此离子筛对溶液中的锂离子有特殊记忆功能,尤其对从海水中提取锂离子具有选择性高、吸附量大的优点,但在吸附理论研究方面还不完善。为此,本文作者用0.5 mol/L HCl对事先用液相法合成的锂离子筛前驱体 LiMn2O4进行酸洗脱锂制得锂吸附剂λ-MnO2,再将离子筛在不同pH和不同Li+浓度溶液中进行吸附,得到离子筛吸附时的动力学和吸附等温线,同时运用XRD和SEM进行相应表征。离子筛λ-MnO2在含锂溶液中对Li+重新吸附,干燥得到吸附Li+后的固体离子筛材料(记为(Li)Mn2O4)。
1 实验部分
1.1 锂离子筛的制备
将一定量制备好的尖晶石型锂离子筛前驱体LiMn2O4加入事先盛有过量(H+相对Li+过量)0.5 mol/L HCl溶液的烧瓶中,于室温(25 ℃)浸渍进行酸浸洗脱,至前驱体中锂基本洗脱完全。酸洗充分后用纯水冲洗3遍,干燥得到锂离子筛,以λ-MnO2表示。
1.2 离子筛吸附性能的研究
将0.2 g离子筛置于50 mL不同pH含有过量Li+的溶液中(LiCl,LiOH和HCl调整pH,溶液中含锂质量浓度约为 250 mg/L,与盐湖卤水中锂质量浓度接近,此时测得所配制LiOH溶液pH为12.95),于25 ℃静态吸附至吸附平衡。取上清液测溶液中锂锰含量,按下式计算锂离子筛对锂离子的吸附容量,研究溶液pH与吸附容量的关系和离子筛的吸附动力学。
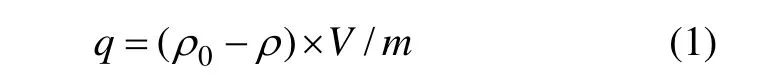
式中:q为每克锂吸附剂吸附 Li+的含量,mg/g;ρ0为Li+起始质量浓度,mg/L;ρ为不同时间的Li+质量浓度,mg/L;V为溶液的体积,L;m为吸附剂的质量,g。
另外,本实验还考察了锂离子浓度对吸附容量的影响。以NaOH调节溶液的pH为12.95,在此pH下配制不同浓度的含锂溶液,研究离子筛吸附容量与Li+浓度的关系及其吸附等温线。
1.3 产物的分析与表征
采用X线衍射仪(Rigaku Dmax 2550型,日本)对离子筛前驱体和离子筛吸附前后的样品进行X线衍射分析,工作参数如下:Cu Kα靶(λ=0.154 056 nm),扫描电压为40 kV,电流为100 mA,步宽为0.02°,扫描速度为 2 (°)/min,扫描范围为10°~85°。用扫描电子显微镜(JSM−6360lv型,日本)观察前驱体和离子筛的微观形貌。采用原子吸收法(TAS−990 F型,北京普析通用仪器有限责任公司制造)测定溶液中锂离子和锰离子的含量。
2 实验结果与讨论
2.1 锂离子筛的制备

图1 离子筛前驱体洗脱效果与酸洗时间的关系Fig.1 Curves of elution effect with elution time
图 1所示为用 0.5 mol/L盐酸对离子筛前驱体LiMn2O4进行酸洗脱锂,锂的迁出率和锰的溶损率随时间变化的情况。由图1(a)可知:前5 h溶液中锂离子浓度几乎呈直线上升,随后趋于平缓;而图1(b)中锰的溶损也在5 h后基本达到平衡。这表明酸洗脱锂过程很快,5 h就能基本完成。由图1可知:酸洗时间越长,洗脱时锂的迁出率则越高,但同时吸附剂中锰的溶损率也越大,5 h酸洗LiMn2O4中锂的迁出率为97%,而锰的溶损率也达到15%。
对离子筛前驱体的酸浸过程,现主要有2种不同的反应机理进行解释:一种是离子交换机理,即认为LiMn2O4在酸浸过程中发生如下离子交换反应[10]:

另一种为氧化还原机理,即Mn3+发生歧化反应[11]:

其中:(□)表示晶格内的空隙,Mn的平均化合价为+3.5。由于Mn3+发生歧化反应,生成Mn4+和Mn2+,Mn4+留在尖晶石骨架中形成λ-MnO2,而Mn2+溶解于溶液中,使Mn的有效离子半径减小。这也在一定程度上解释 LiMn2O4在 Li+迁出后其尖晶石结构不变,而只是晶胞常数略减小的原因。
2.2 吸附性能的研究
2.2.1 吸附容量与溶液pH的关系
由实验得到锂吸附饱和容量随 pH的变化关系如图2所示。由图2可以看出:在酸性溶液中λ-MnO2对Li+离子的吸附极其微弱,甚至在pH<3时,离子筛没有对Li+没有吸附,反而因pH低使离子筛中残留的锂有少量迁出,在中性溶液中,λ-MnO2对Li+的吸附仍然很少,当溶液pH=7.13时,吸附容量只有 0.9 mg/g。随着溶液碱性的增加,λ-MnO2对 Li+的吸附能力逐渐增强;当pH<11时,吸附容量随pH的增大呈缓慢增加趋势,当pH>11时,吸附容量发生了突变,由2.2 mg/g增加到pH为12.95时的23.75 mg/g,但这与λ-MnO2的理论吸附容量38.4 mg/g仍有一定差距。

图2 离子筛吸附容量与溶液pH的关系Fig.2 Relationship between adsorptive capacity and pH
钟辉等[12]认为在离子筛的吸附中心具有酸式解离能力的OH基团,可与Li+按等量交换反应进行,即:

离子筛的官能基团具有三元酸的性质,最后一类OH基团只有在pH>11.5时才能参加离子交换反应。基于这种机理,在锂溶液酸性较大时,不利于离子筛的解离,因而,此时离子筛的交换容量较低,即锂的吸附容量较低;随着溶液 pH的增大,离子筛的解离度增大,因而,其对锂的吸附能力增大。
由于离子筛λ-MnO2对Li+的吸附是释放H+、消耗OH−的过程,随着溶液 pH的增大,反应向正方向进行,因此,离子筛对 Li+的吸附容量随 pH的增加而增大。
2.2.2 吸附容量与吸附时间的关系及其吸附动力学
将一定量的离子筛置于pH为12.95的LiOH溶液中,并于25 ℃水浴加热,间隔一定时间取上清液分析离子筛对锂的吸附容量,得到如图3所示的曲线。由图3可知:离子筛的吸附交换速率较快,在较短时间内就能达到平衡,吸附1 h锂的吸附容量为17.8 mg/g,到 24 h基本达到平衡,48 h锂的吸附容量为 23.75 mg/g。

图3 离子筛吸附容量与吸附时间的关系Fig.3 Relationship between adsorptive capacity and time of adsorption
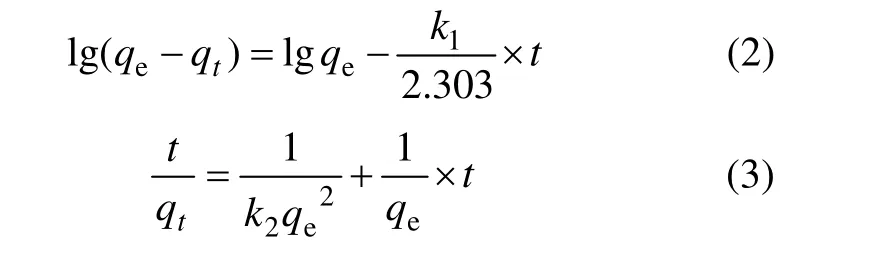
吸附剂对金属离子的吸附动力学研究已有不少报道,目前伪一级动力学模型(式(2))和伪二级动力学模型(式(3))被广泛应用来研究吸附机理[13−14],确定吸附过程的速率常数。式中:qe和qt分别为在吸附平衡和吸附时间为t时吸附剂对 Li+的吸附容量,mg/g;k1和k2分别为伪一级动力学模型和伪二级动力学模型吸附速率常数,单位分别为 h−1和 g·mg−1·h−1。

图4 LiOH溶液中锂吸附的伪一级动力学和伪二级动力学曲线Fig.4 Pseudo-first-order and pseudo-second-order kinetic plots for Li+ adsorption in LiOH solution

表1 LiOH溶液中锂吸附的动力学参数Table 1 Kinetic parameters for Li+ adsorption in LiOHsolution
用伪一级动力学方程和伪二级动力学方程对图 3中曲线进行线性拟合,结果如图4和表1所示。由图4和表1可以看出:用伪二级动力学方程拟合的线性相关系数比伪一级动力学方程拟合的大,达到0.999 4。所以认为吸附剂在LiOH溶液中的吸附动力学符合伪二级动力学模型,根据伪二级动力学方程,求出反应的速率常数为 0.050 9 g·mg−1·h−1,平衡吸附容量分别为24.39 mg/g,此吸附容量与图3中实验得到的平衡吸附容量23.75 mg/g相近。这进一步说明吸附剂在LiOH溶液中的吸附符合伪二级动力学模型,表明吸附过程主要为化学吸附,吸附剂对锂离子的吸附为单分子层吸附[15]。
2.2.3 吸附容量与Li+浓度的关系及其吸附等温线
以NaOH调节溶液的pH为12.95,在此pH下配制不同浓度的含锂溶液,得到离子筛吸附容量与 Li+浓度的关系如图5所示。由图5可知:Li+浓度对离子筛吸附容量有一定的影响。当Li+浓度为0.02 mol/L时离子筛的吸附容量为 21.8 mg/g;而当 Li+浓度为 0.1 mol/L时,离子筛的吸附容量为24.16 mg/g。

图5 吸附容量与Li+浓度的关系Fig.5 Relationship between adsorptive capacity and Li+ concentration
锂吸附剂对Li+的吸附可以通过Langmuir吸附等温方程(式(4))和 Freundlich吸附等温方程(式(5))拟合[16]。

式中:ρe为吸附平衡后溶液中Li+的质量浓度,mg/L;qe为平衡吸附容量,mg/g;qm为最大吸附容量,mg/g;KL为Langmuir实验常数,L/mg;n和KF为与吸附容量和吸附速率有关的Freundlich常数。
分别用Langmuir吸附等温方程和Freundlich吸附等温方程对离子筛在不同浓度的含锂溶液中平衡时的浓度和吸附容量进行拟合,结果如图6和表2所示。从图6和表2可以看出:用Langmuir吸附等温方程进行拟合的线性关系比Freundlich拟合的好,线性相关系数达到0.990 4,表明Li+的吸附符合Langmuir吸附等温方程,最大吸附容量为24.29 mg/g,与图5中在Li+浓度为0.1 mol/L时离子筛的吸附容量24.16 mg/g相近。

图6 Langmuir和Freundlich等温线Fig.6 Langmuir and Freundlich isotherms curves

表2 锂吸附剂的等温吸附模型及参数Table 2 Type and parameters of isothermal adsorption of Li-adsorbent
2.3 离子筛吸附Li+前后的结构和形貌
锂离子筛前驱体经过酸洗所得酸洗后物理论上就是锂离子筛(又称锂吸附剂)。这种锂离子筛在某种理论上可认为是锂离子被氢离子完全替代后的尖晶石结构锰氧化物,可简写为λ-MnO2。而根据离子交换机理,吸附Li+后的离子筛理论上又替代H+的位置,转化为LiMn2O4。对此选取前驱体及其离子筛吸附前后的样品,用X线衍射对其进行结构分析,确定其与标准的λ-MnO2晶体结构是否存在差别,最后用扫描电镜分析其外部形貌和结晶情况相对于其前驱体是否发生了变化。离子筛前驱体及其离子筛吸附前后的 XRD图谱和SEM照片分别如图7和图8所示。

图 7 LiMn2O4,λ-MnO2和(Li)Mn2O4的XRD图谱Fig.7 XRD patterns of LiMn2O4, λ-MnO2 and (Li)Mn2O4

图8 LiMn2O4,λ-MnO2和(Li)Mn2O4的SEM照片Fig.8 SEM images of LiMn2O4, λ-MnO2 and (Li)Mn2O4
由图7可知:酸浸洗脱后得到产物结构与酸浸前LiMn2O4的相似,仍保持尖晶石结构,各特征衍射峰所对应的角 2θ对应于前驱体 LiMn2O4向高角度方向移动。这说明盐酸对LiMn2O4的酸浸过程中,随着Li+的迁出,H+的迁入,使晶胞发生了收缩,晶胞常数a有所减小,形成了细微的空腔。这是H+半径小于Li+半径所致。这是离子筛具有筛效应而能够吸附锂离子的微观原因。对照标准XRD图谱,离子筛吸附锂后,其图谱与离子筛的图谱比较,各特征峰数目未变,其峰高和峰间距也没发生明显的改变,这说明离子筛在吸附锂后其产物的尖晶石晶体骨架结构未发生变化,只是各特征峰的位置对应于离子筛有向低角度移动。可见:在离子筛吸附锂的过程中,随着交换反应的进行,原先H+的位置又被Li+取代;离子筛吸附过程中,由锂离子替代氢离子后其晶胞又扩大,从而与前驱体的晶体结构基本一致。这在一定程度上也说明锂离子筛的吸附交换是成功的。
由图8可以看出:产物是粒径在0.5~2 μm范围内的粉末材料,颗粒之间较为松散,这是由于焙烧过程中放出CO2和O2,使颗粒内部产生较多的孔隙,从而形成联通的网络结构,这有利于离子在结构中的迁出和嵌入。离子筛前驱体颗粒不够均匀,但粒子团聚严重。离子筛吸附后的颗粒则比较均匀,晶粒发育更为成熟,颗粒粒度较吸附前要小,但仍有部分团聚。
4 结论
(1) 锂离子的迁出和锰的溶损与洗脱的时间有关。洗脱时间越长,锂的迁出率则越高,但同时锰的溶损率也高。
(2) 离子筛对Li+的吸附容量随着pH升高而增大,pH为12.95的LiOH溶液中离子筛的吸附容量为23.75 mg/g。
(3) 吸附过程符合伪二级动力学方程和 Langmuir等温吸附方程。吸附过程主要为化学吸附,对锂离子的吸附为单分子层吸附。
(4) 锂离子筛吸附交换前后晶体结构只发生了细微变化,但都是尖晶石结构,离子筛吸附锂后与前驱体的晶体结构基本一致。
[1] Hamzaoui A.H, M′nif A, Hammi H, et al. Contribution to the lithium recovery from brine[J]. Desalination, 2003, 158(1/3):221−224.
[2] Panero S, Ciuffa F, D'Epifano A, et al. New concepts for the development of lithium and proton conducting membranes[J].Electrochimica Acta, 2003, 48(14/16): 2009−2014.
[3] Jannik J, Maier J. Nanocrystallinity effects in lithium battery materials. Aspects of nano-ionics. Part IV[J]. Physical Chemistry Chemical Physics, 2003, 5(2/3): 5215−5220.
[4] 董殿权, 张凤宝, 张国亮, 等. LiMg0.5Mn1.5O4的合成及对 Li+的离子交换选择性[J]. 无机化学学报, 2004, 20(9): 1126−1130.DONG Dian-quan, ZHANG Feng-bao, ZHANG Guo-liang, et al.LiMg0.5Mn1.5O4synthesis and its selectivity to Li+exchange[J].Chinese Journal of Inorganic Chemistry, 2004, 20(9):1126−1130.
[5] Koyanaka H, Matsubaya O, Koyanaka Y, et al. Quantitative correlation between Li adsorption and H content in manganese oxide spinelλ-MnO2[J]. Journal of Electroanalytical Chemistry,2003, 559(15): 77−81.
[6] Chitrakar R, Kanoh H, Miyai Y, et al. A new type of manganese oxide (MnO2·0.5H2O) derived from Li1.6Mn1.6O4and its lithium ion-sieve properties[J]. Chemistry of Materials, 2000, 12(10):3152−3157.
[7] ZHANG Yong-cai, WANG Hao, WANG Bo, et al. Low temperature synthesis of nanocrystalline Li4Mn5O12by a hydrothermal method materials[J]. Research Bulletin, 2002,37(8): 1411−1417.
[8] LIU Zhan-qiang, WANG Wen-lou, LIU Xian-ming, et al.Synthesis of nanostructured spinel LiMnO by hydrothermal method at 70 ℃ [J]. Inorganic Chemistry Communications, 2004,7(2): 308−310.
[9] Myung S T, Komaba S, Kumagai N. Hydrothermal synthesis and electrochemical behavior of orthorhombic LiMnO2[J].Electrochimica Acta, 2002, 47(20): 3287−3295.
[10] Clearfield A. Inorganic ion exchangers, past, present and future[J]. Solvent Extraction and Ion Exchange, 2000, 18(4):655−678.
[11] Huter J C. Preparation of a new crystal form of manganese dioxideλ-MnO2[J]. Journal of Solid State Chemistry, 1981, 39(2):142−147.
[12] 钟辉, 殷辉安. 偏钛酸型离子交换剂表面性质与选择吸附性研究[J]. 离子交换与吸附, 2003, 19(1): 55−60.ZHONG Hui, YIN Hui-an. Study on the properties of the surface and absorb of Li+ion-exchange of H2TiO3type[J]. Ion Exchange and Adsorption, 2003, 19(1): 55−60.
[13] Barkat M, Nibou D, Chegrouche S, et al. Kinetics and thermodynamics studies of chromium(VI) ions adsorption onto activated carbon from aqueous solutions[J]. Chemical Engineering and Processing, 2009, 48(1): 38−47.
[14] Benhammou A, Yaacoubi A, Nibou L, et al. Adsorption of metal ions onto Moroccan stevensite: Kinetic and isotherm studies[J].Colloid and Interface Science, 2005, 282(2): 320−326.
[15] Naiya T K, Bhattacharya A K, Das S K. Removal of Cd (II) from aqueous solutions using clarif i ed sludge[J]. Colloid and Interface Science, 2008, 325(1): 48−56.
[16] WANG Lu, MENG Chang-gong, HAN Mei, et al. Lithium uptake in fixed-pH solution by ion sieves[J]. Colloid and Interface Science, 2008, 325(1): 31−40.
猜你喜欢
山东冶金(2022年4期)2022-09-14 08:58:10
耐火材料(2022年4期)2022-08-28 03:01:10
中国宝玉石(2022年2期)2022-04-25 06:37:16
科学(2020年1期)2020-08-24 08:07:56
中国有色金属学报(2018年2期)2018-03-26 07:58:38
材料科学与工程学报(2016年1期)2017-01-15 13:33:52
当代化工研究(2016年7期)2016-03-20 16:21:54
电源技术(2015年9期)2015-06-05 09:36:06
电源技术(2015年9期)2015-06-05 09:36:04
应用化工(2014年11期)2014-08-16 15:59:13
