
细菌−矿物接触/非接触模式下黄铜矿浸出溶解行为
2011-09-17 07:34:30顾帼华郭玉武
中南大学学报(自然科学版) 2011年8期
顾帼华,郭玉武,
(1. 中南大学 资源加工与生物工程学院,湖南 长沙,410083;2. 湖南有色金属研究院,湖南 长沙,410015)
黄铜矿是自然界中最主要的铜矿物,也是最难浸出的矿物之一[1]。研究者们围绕细菌对黄铜矿浸出作用的机理开展了大量的研究工作并提出了一些理论,其中直接作用说和间接作用说机理占据主导地位[2−3]。直接作用机理是指细菌吸附在矿物表面,直接以硫化矿为能源物质,通过自身的酶将硫化矿分解成硫酸盐并将硫化矿中的硫直接氧化成硫酸[4]。间接作用机理认为:硫化矿的浸出是通过Fe3+的氧化作用完成的,其中产生了Fe2+和元素硫,细菌在浸出中的作用是将Fe2+氧化成 Fe3+和将元素硫氧化成硫酸[5]。Rodriguez等[6]认为细菌通过间接作用对黄铜矿进行浸出,而傅建华等[7]则认为吸附在矿物表面的细菌通过改变矿物表面化学性质来影响黄铜矿的浸出,黄铜矿生物浸出以直接作用为主。可见,细菌对黄铜矿浸出作用机理仍存在较大争论。在提高黄铜矿生物浸出效率方面,研究者也开展了大量工作。由于黄铜矿生物浸出是一个电化学腐蚀过程,浸出体系氧化还原电位也对浸出至关重要[8−9]。Third等[10]的研究表明浸出体系电位对黄铜矿浸出的影响远大于细菌数量或活性对其的影响,认为适宜的电位更有利于黄铜矿的浸出。研究认为导致黄铜矿浸出速率低的原因也是由于在浸出过程中矿物表面形成了钝化膜,但人们对钝化膜的性质仍存在争议。Dutrizac[11]通过SEM在黄铜矿表面观测到一层致密的硫膜,并测定了黄铜矿在不同试验条件下的浸出速率,认为是硫膜阻碍了黄铜矿的浸出。在细菌−矿物接触情况下,吸附在黄铜矿表面的细菌可以将硫膜氧化,从而消除了黄铜矿表面的钝化膜,促进了黄铜矿的进一步浸出。Watling等[12−13]认为浸出体系生成的黄钾铁矾沉淀会覆盖在黄铜矿表面并阻碍细菌和氧化物与矿物表面的作用以及表面产物的扩散,从而抑制了黄铜矿的进一步浸出,因此,认为黄钾铁矾沉淀是导致黄铜矿表面钝化和浸出率低的主要原因。到目前为止,研究者对细菌在黄铜矿生物浸出中的作用机理和引起黄铜矿表面钝化的原因仍未达成一致观点。在此,本文作者通过利用透析袋阻止细菌和矿物的直接接触,研究不同浸出阶段细菌−矿物接触/非接触模式对黄铜矿浸出的影响,并通过对浸渣的SEM和XRD检测,探讨不同浸出阶段细菌浸出黄铜矿机制及黄铜矿表面钝化的原因。
1 材料和方法
1.1 微生物
试验所需嗜酸氧化亚铁硫杆菌(ATCC23270)由中南大学生物冶金重点实验室提供。在起始pH为2.0,温度为30 ℃条件下,嗜酸氧化亚铁硫杆菌经含3%黄铜矿的0K培养基转移驯化3月,以适应黄铜矿为其能源物质。0 K培养基[14]成分为:(NH4)4SO43.0 g/L,KCl 0.1 g/L,K2HPO40.5 g/L,MgSO4·7H2O 0.5 g/L,Ca(NO3)20.01 g/L。
1.2 矿物
试验所需黄铜矿来自中国湖北大冶铜矿,矿样经手工挑杂后,再经瓷球磨矿、干式筛分至粒度低于75 μm,用于浸出试验。对纯矿物样品成分进行化学分析,各元素质量分数为:32.51% Cu,31.92% Fe,33.47% S,纯度达到94.5%。
1.3 浸出试验
设计以下3组试验:(1) 细菌−矿物直接接触试验(A1);(2)细菌−矿物透析袋隔离试验(A2),透析袋每隔3 d更换1次;(3) 细菌−矿物透析袋隔离试验(A3),在试验过程中不更换透析袋。对于试验A2和A3,透析袋材质为聚偏氟乙烯,截留相对分子质量为8 000,使用前先用酒精浸泡8 h,再经1 mmol/L盐酸浸泡3 h,最后用去离子水反复清洗后待用,浸出试验时将3.6 g黄铜矿用透析袋包裹并密封后放入摇瓶中,透析袋阻碍细菌与矿物的接触。3组浸矿试验条件如下:在250 mL锥形瓶中加入120 mL 0K培养基溶液,接菌量(体积分数)为10%,矿浆体积分数为3%,摇床转速为170 r/min,温度为 30 ℃。驯化后的嗜酸氧化亚铁硫杆菌用黄铜矿矿浆体积分数为3%的0K培养基培养至对数期,后经滤纸(d=5 μm)过滤和离心收集细菌(转速为10 000 r/min)。收集到的细菌用经硫酸调pH=2.0的蒸馏水清洗2遍后保存在0K培养液中,并对浸出试验进行接种。接种后溶液中的细菌浓度为4×107个/mL。在浸出试验过程中,挥发的水分用蒸馏水补加。
1.4 分析方法
在浸出试验期间,浸出液经取样后用原子吸收光谱法测定其中Cu2+质量浓度,用重铬酸钾溶液氧化滴定其中Fe3+质量浓度。浸出液氧化还原电位用铂电极来测量(以饱和甘汞电极为参比电极)。浸出液pH用酸度计测量,酸度计每次使用前都校准。在浸出过程中,细菌用光学显微镜计数,细菌活性通过其氧化Fe2+速率来间接测得。浸渣经过滤后冷冻干燥,分别进行SEM和XRD检测。
2 结果与讨论
2.1 浸出过程中细菌浓度和活性的变化
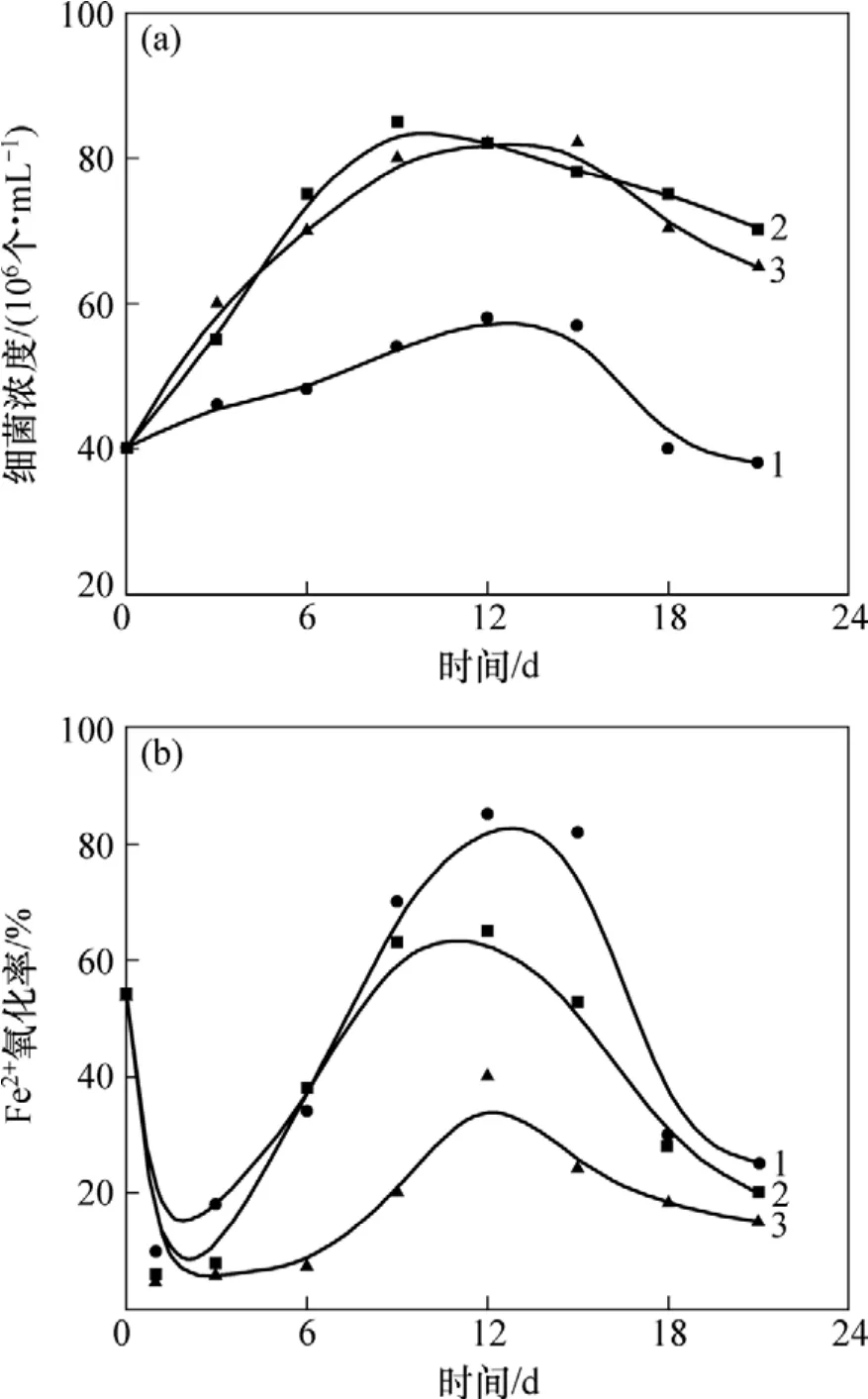
图1 浸出试验体系细菌数目和活性Fig.1 Number and activity of bacteria in leaching experiments
在浸出试验过程中,体系细菌数目和活性变化如图1所示,其中,细菌活性是指其氧化Fe2+的能力。由图1可知:试验A1,A2和A3中细菌浓度和活性在浸出前期一直上升,从第15天起开始下降。图1(a)中,试验A1中的细菌浓度明显比A2和A3中的小,其主要原因是 A1中的细菌大量吸附在矿物表面,从而导致浸出体系游离细菌浓度减少。由图1(b)可知:试验A1,A2和A3中细菌活性都随浸出时间的延长而先升高后下降,在12 d左右活性达到最高,且A1中细菌活性比试验A2和A3的高,表明细菌与黄铜矿直接接触时可以获得更高的活性。在浸出过程中,试验A2和A3透析袋表面有黄色沉淀物生成,且沉淀数量随着浸出时间的延长而增加。该黄色沉淀物经XRD检测为黄钾铁矾(图5)。由于试验A3中没有更换透析袋,故随着浸出试验的进行,透析袋表面形成了一层黄钾铁矾沉淀膜,透析袋内外离子交换受到抑制,从而导致A3中细菌活性比A2的低。从第15天开始,由于黄钾铁矾沉淀开始大量生成,细菌生存环境受到恶化,故此时试验A1,A2和A3中细菌活性都开始急剧下降。
2.2 浸出过程中离子质量浓度的变化
在浸出试验过程中,体系Cu2+和Fe3+质量浓度变化如图2(a)和2(b)所示。由图2(a)和2(b)可知:
在第1阶段(0~3 d),在试验A1,A2和A3中,浸出体系Cu2+质量浓度基本相同,分别为0.21,0.20和0.18 g/L;试验A1体系中Fe3+质量浓度为0.33 g/L,略高于试验A2(0.29 g/L)和试验A3(0.32 g/L)。
在第2阶段(3~12 d),在试验A1和A2中,Cu2+和Fe3+质量浓度都继续上升,但随着浸出的进行,上升速率亦都有所下降。浸出12 d后,在试验A1中,Cu2+和Fe3+质量浓度分别为0.43 g/L和0.62 g/L;在试验A2中,Cu2+和Fe3+质量浓度浸出12 d后分别为0.37 g/L和0.38 g/L。在试验A1和A2中,Cu2+质量浓度差异较小。与试验A1和A2相比,试验A3中Cu2+质量浓度上升得较缓慢,体系中Fe3+质量浓度也开始下降,浸出12 d后,Cu2+和Fe3+质量浓度也都比试验A1和A2的低。
在第3阶段(12~21 d),试验A1和A3中Cu2+和Fe3+质量浓度基本不变,试验 A2中 Cu2+质量浓度不变,但Fe3+质量浓度下降。浸出21 d后,试验A1,A2和 A3体系中 Cu2+质量浓度分别为 0.51,0.43和0.32 g/L;Fe3+质量浓度分别为0.70,0.10和0.11 g/L。
2.3 浸出过程体系电位和pH的变化
在浸出试验过程中,体系电位和pH变化如图2(c)和2(d)所示。由图2(c)和2(d)可知:
在第1阶段(0~3 d),试验A1,A2和A3中体系氧化还原电位都上升,且上升幅度基本相同;试验A1,A2和A3中体系pH都有所上升,但A1比A2和A3的上升幅度小。
在第2阶段(3~12 d),试验A1和A2中体系氧化还原电位都继续上升,但A1上升较快,从535 mV升至568 mV,A2仅由525 mV升至537 mV,变化不明显;试验A3体系氧化还原电位却急剧下降,从534 mV降为436 mV。试验A1体系氧化还原电位比A2和A3的高。试验A1中体系pH开始下降,而试验A2和A3中体系pH却一直维持在2.6左右。
第3阶段(12~21 d),试验A1中体系电位继续上升,pH继续下降,浸出21 d后,体系电位和pH分别为615 mV和1.8;试验A2中体系电位和pH都有所下降,浸出21 d后,体系电位和pH分别为485 mV和2.4;试验A3中体系电位和pH基本维持在425 mV和2.7左右不变。

图2 浸出试验结果Fig.2 Leaching experiments results
2.4 分析与讨论
在黄铜矿浸出过程中,主要发生如下反应:

由式(1)可知:黄铜矿浸出过程中优先释放 Fe3+,即黄铜矿表面铁比铜更易浸出,故在第1阶段(0~3 d),试验A1,A2和A3中Fe3+质量浓度都要高于Cu2+的质量浓度。黄铜矿分解过程中有铜蓝和单质硫生成,且铜蓝在低电位下不易发生分解。细菌的加入可以提高浸出体系电位,使铜蓝进一步发生分解并释放Cu2+。
在第1阶段(0~3 d),试验A1,A2和A3体系氧化还原电位上升,是细菌将Fe2+氧化为Fe3+所致,而试验A1,A2和A3中pH的上升主要是酸耗所致。对于试验A1,因吸附在矿物表面的细菌可以将反应生成的单质硫氧化为硫酸而补偿酸耗,故其 pH上升幅度比实验A2和A3的上升幅度小。试验A1,A2和A3中体系Cu2+和Fe3+质量浓度都基本相同,表明细菌与矿物接触与否对黄铜矿浸出并无影响,细菌主要通过将 Fe2+氧化为 Fe3+而提高体系氧化还原电位来促进Cu2+的浸出,因此,可以认为该阶段黄铜矿浸出主要以Fe3+氧化浸出为主。
在第2阶段(3~12 d),试验A1中体系pH下降的主要原因是该阶段吸附在黄铜矿表面的细菌将矿物表面生成的硫氧化为硫酸。试验 A1体系氧化还原电位比试验A2和A3的高,表明细菌−矿物接触模式与非接触模式相比,接触模式下更有利于提高浸出体系氧化还原电位中。高电位下黄铜矿更易发生氧化溶解,故试验A1 Cu2+质量浓度比试验A2和A3的高。试验A3透析袋表面开始有黄钾铁矾沉淀生成。Fe3+生成黄钾铁矾沉淀使体系 Fe3+质量浓度降低是导致试验 A3体系氧化还原电位下降的主要原因,反应如式(5)所示。对于试验A2,由于每隔3 d更换透析袋,故避免了黄钾铁矾沉淀在透析袋表面覆盖。尽管吸附在矿物表面的细菌将矿物表面的硫膜氧化消除,但该阶段试验A1和A2中Cu2+质量浓度差异较小,表明矿物表面生成的硫膜对黄铜矿浸出无明显抑制作用,这与Cordoba等[15]的研究结论一致。通过分析比较试验A2和 A3浸出试验结果,认为黄钾铁矾沉淀是导致试验A3体系Cu2+质量浓度和细菌活性较低的主要原因。
在第3阶段(12~21 d),试验A1溶液中开始有大量黄钾铁矾沉淀生成,尽管试验A2中每隔3 d更换1次透析袋,但透析袋表面仍有黄钾铁矾沉淀堆积。在高电位下,浸出体系易生成大量黄钾铁矾沉淀。黄钾铁矾沉淀是引起试验A2中Fe3+质量浓度和pH变化的主要原因。由图3可知:黄钾铁矾沉淀大量覆盖于矿物表面;黄钾铁矾沉淀使黄铜矿表面严重钝化,从而抑制黄铜矿的进一步浸出,故该阶段试验 A1和 A2体系中Cu2+质量浓度基本不变,黄铜矿浸出都受到显著抑制。对于试验A3,由于第2阶段(3~12 d)中透析袋表面生成的黄钾铁矾沉淀膜基本抑制了黄铜矿的进一步浸出,故在第3阶段(12~21 d),试验A3体系中各因素均无明显变化。黄钾铁矾沉淀的生成亦会恶化细菌生存环境,导致试验A1和 A2中细菌活性急剧下降。
2.5 浸渣表面分析
试验A1和A2中,浸出21 d的浸渣表面SEM和XRD检测结果分别如图3和图4所示。试验A1中,黄铜矿浸渣表面不仅腐蚀严重,而且覆盖有大量黄钾铁矾沉淀,表面受到严重钝化。试验 A2中,黄铜矿浸渣表面也有明显的浸蚀和少量黄钾铁矾沉淀。
浸出21 d后,试验A2透析袋表面沉淀物经SEM和XRD检测,结果分别如图5所示。由图5可知:该沉淀物为黄钾铁矾。黄钾铁矾沉淀在黄铜矿表面的大量覆盖阻碍了浸出液与黄铜矿表面接触或透析袋内外物质交换,从而使黄铜矿浸出速率下降。因此,可以认为试验A1和试验A2中黄铜矿浸出率下降主要是由黄钾铁矾沉淀引起的。黄钾铁矾沉淀是导致黄铜矿表面钝化和浸出率低的主要原因。

图3 浸出21 d后黄铜矿浸渣表面SEMFig.3 SEM micrographs of the chalcopyrite surfaces after 21 d of leaching

图4 浸出21 d后黄铜矿浸渣XRD图Fig.4 XRD patterns of chalcopyrite residues after 21 d leaching

图5 浸出21 d后A2透析袋表面沉淀SEM和XRD图Fig.5 SEM micrograph and XRD of precipitations on dialysis bag surface of A2 after 21 d leaching
3 结论
(1) 在细菌−矿物接触模式下,细菌通过氧化矿物表面硫及氧化Fe2+生成Fe3+来促进黄铜矿浸出;而矿物−细菌非接触模式下,细菌氧化Fe2+产生的Fe3+在黄铜矿氧化溶解中起主要作用。
(2) 浸出体系电位是影响黄铜矿生物浸出的主要因素。细菌−矿物接触模式比非接触模式更有利于提高浸出体系电位,并使黄铜矿表面生成的硫膜经氧化消除,因而更有利于黄铜矿的浸出。
(3) 在细菌−矿物接触和非接触模式下,易于在较高电位下生成的黄钾铁矾沉淀是导致黄铜矿表面钝化及抑制黄铜矿进一步浸出的主要原因。
[1] Dutrizac J E. The kinetics of dissolution of chalcopyrite in ferric iron media[J]. Metallurgical and Materials Transactions B, 1978,9(4): 431−439.
[2] Cordoba E M, Munoz J A, Blazquez M L, et al. Leaching of chalcopyrite with ferric ion. Part Ⅰ: General aspects[J].Hydrometallurgy, 2008, 93(3/4): 81−87.
[3] Mikhlin Y L, Tomashevivh Y V, Asanov I P, et al. Spectroscopy and electrochemical characterization of the surface layers of chalcopyrite (CuFeS2) reacted in acidic solutions[J]. Applied Surface Science, 2004, 225(1/2/3/4): 395−409.
[4] Silverman M P, Ehrlich H L. Microbial formation and degradation of minerals[J]. Adv Appl Microbiol, 1964, 6(1):153−206.
[5] Sand W, Gehrke T, Hallmann R, et al. Sulfur chemistry, biofilm and the indirect attract mechanism: A critical evaluation of bacterial leaching[J]. Appl Microbiol Biotechnol, 1995, 43(1):961−966.
[6] Rodriguez Y, Ballester A, Blazquez M L, et al. New information on the chalcopyrite bioleaching mechanism at low and high temperature[J]. Hydrometallurgy, 2003, 71(1): 47−56.
[7] 傅建华, 邱冠周, 胡岳华, 等. 浸矿细菌的超微结构及其特性[J]. 中南大学学报: 自然科学版, 2004, 35(4): 562−568.FU Jian-hua, QIU Guan-zhou, HU Yue-hua, et al. Ultrastructure and properties of leaching bacteria[J]. Journal of Central South University: Science and Technology, 2004, 35(4): 562−568.
[8] Cordoba E M, Munoz J A, Blazquez M L, et al. Leaching of chalcopyrite with ferric ion. Part Ⅳ: The role of redox potential in the presence of mesophilic and thermophilic bacteria[J].Hydrometallurgy, 2008, 93(3/4): 106−115.
[9] Pradhan N, Nathasharma K C, Rao K S, et al. Heap bioleaching of chalcopyrite: A review[J]. Minerals Engineering, 2008, 21(5):355−365.
[10] Third K A, Cord-Ruwisch R, Watling H R. The role of iron-oxidizing bacteria in stimulation or inhibition of chalcopyrite bioleaching[J]. Hydrometallurgy, 2000, 53(3):225−233.
[11] Dutrizac J E. Elemental sulphur formation during the ferric sulphate leaching of chalcopyrite[J]. Can Metall Q, 1989, 28(4):337−344.
[12] Watling H R. The bioleaching of sulphide minerals with emphasis on copper sulphides: A review[J]. Hydrometallurgy,2006, 84(1): 81−108.
[13] Sandstroma A, Shchukarevb A, Paul J. XPS characterisation of chalcopyrite chemically and bio-leached at high and low redox potential[J]. Minerals Engineering, 2005, 18(5): 505−515.
[14] Silveman M P, Lundgren D G. Studies on the chemoautotrophic iron bacteriumFerrobacillus ferrooxidans[J]. J Bacteriol, 1959,77(5): 642−647.
[15] Cordoba E M, Munoz J A, Blazquez M L, et al. Leaching of chalcopyrite with ferric ion. Part Ⅱ: Effect of redox potential[J].Hydrometallurgy, 2008, 93(3/4): 88−96.
猜你喜欢
矿产综合利用(2022年1期)2022-03-30 11:35:24
小哥白尼(趣味科学)(2021年10期)2022-01-17 02:42:14
金属矿山(2020年10期)2020-11-14 11:20:40
学生天地(2020年10期)2020-08-25 09:14:34
矿产综合利用(2020年1期)2020-07-24 08:51:28
山东国资(2020年4期)2020-06-10 09:14:48
金属矿山(2018年12期)2019-01-14 08:26:36
中成药(2018年2期)2018-05-09 07:19:55
科技知识动漫(2017年12期)2018-03-07 16:46:23
四川地质学报(2017年1期)2017-06-15 20:28:58
