
反相高效制备液相色谱法制备洋川芎内酯H和I
2011-08-24 00:55:52张翠萍李行诺陈鸳谊葛志伟颜继忠
浙江工业大学学报 2011年4期
张翠萍,李行诺,陈鸳谊,葛志伟,颜继忠
(1.浙江工业大学 药学院,浙江杭州 310032;2.浙江大学 药物信息学研究所,浙江杭州 310058)
反相高效制备液相色谱法制备洋川芎内酯H和I
张翠萍1,李行诺1,陈鸳谊1,葛志伟2,颜继忠1
(1.浙江工业大学 药学院,浙江杭州 310032;2.浙江大学 药物信息学研究所,浙江杭州 310058)
从川芎中分离制备洋川芎内酯H和洋川芎内酯I对照品.用8倍量80%乙醇回流提取川芎药材,得到川芎浸膏经D-101大孔吸附树脂、硅胶柱色谱及反相高效制备液相色谱分离,获得两个化合物单体.经核磁共振波谱分析,确定其分别为洋川芎内酯H和洋川芎内酯I,两者纯度均高于98%,可作为对照品进行定量分析.该方法操作简单,重现性好,上样量大,能够同时获得大量的高纯度的目标成分,为洋川芎内酯H和洋川芎内酯I单体的制备研究提供了一种新的方法.
反相高效制备液相色谱法;川芎;洋川芎内酯H;洋川芎内酯I
洋川芎内酯H和洋川芎内酯I是一对结构相似的苯酞类化合物(图1),在中药川芎(Ligusticum chuanxiongHort)中含量较大[1].现代药理研究发现,二者对于心血管和肿瘤疾病具有较好的疗效[2-7].但二者化学性质不稳定,在高温和碱性条件下易发生分解、缩合等反应[8-9].采用传统的分离纯化方法,操作繁杂,分离效率低,难以获得二者高纯度的单体.
高效制备液相色谱法不受样品挥发性和热稳定性的限制,分离效能高、分析速度快、检测灵敏,操作简单,上样量大,重现性好,适合分离纯化不稳定的天然产物.本实验使用反相高效制备液相色谱并结合D-101大孔吸附树脂、开放硅胶柱色谱,从中药川芎中同时分离制备洋川芎内酯H和洋川芎内酯I,为建立川芎药材及其制剂的质量控制标准奠定了基础.
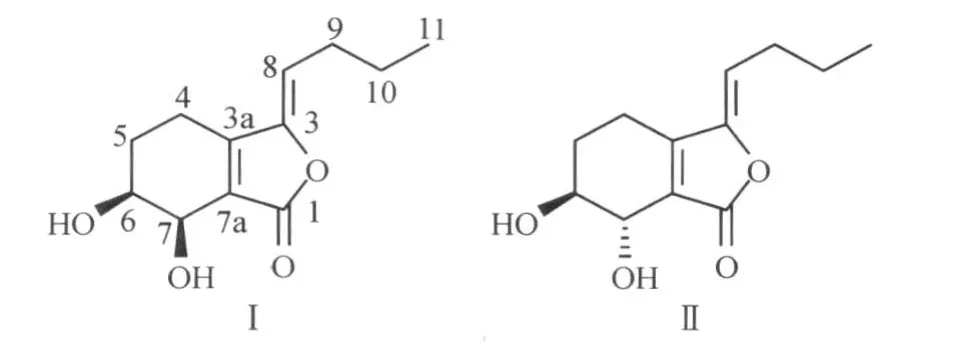
图1 洋川芎内酯H(Ⅰ)和洋川芎内酯I(Ⅱ)的结构式Fig.1 Structures o f senkyuno lide H(Ⅰ)and senkyunolide I(Ⅱ)
1 实验部分
1.1 仪器与试剂
Shimadzu LC-8A高效制备液相色谱仪(日本岛津制作所),包括SPD-M 20A diode array Detector,LC-8ADvp两元泵,LCsolution色谱数据工作站;Lum tech(绿绵)分析型高效液相色谱仪;M ini spin离心机(Eppendorf公司);U ltrasonic Generator(型号H 66MC,无锡超声电子设备有限公司);梅特勒托利多A L104电子天平(瑞士 Mettler To ledo);Bruker AC-80核磁共振仪(NMR);岛津 UV 2487紫外光谱仪;Eyela旋转蒸发仪;M illi-Q纯净水;100-200目硅胶(青岛海洋化工厂);硅胶GF254由青岛海洋化工厂生产;D-101型大孔吸附树脂(安徽三星树脂科技有限公司);色谱甲醇(江苏汉邦科技有限公司);石油醚(60~90℃)、乙酸乙酯和乙醇均为分析纯;川芎药材购于杭州胡庆余堂,样品保存于浙江工业大学药学院.
1.2 样品前处理
取川芎药材3 kg,粉碎成粗粉,8倍量80%乙醇回流提取2次,合并提取液,减压浓缩成浸膏750 mL,加适量纯净水混合分散至1450 mL,加到处理好的D101大孔吸附树脂(1300 g,φ10 cm ×100 cm)中,静止吸附6 h,用水洗至还原糖呈中性(M olish反应),分别用50%和90%的乙醇溶液洗脱大孔吸附树脂,并收集洗脱液,减压浓缩.得到50%乙醇洗脱组分26 g,90%乙醇洗脱组分65.6 g.洋川芎内酯H和洋川芎内酯I结构中含有两个羟基,极性较大,因此,主要集中在50%乙醇洗脱组分中.将50%乙醇洗脱组分干膏26 g用乙醇溶解拌入硅胶(100~200目)55 g,减压旋干,干法上样,使用正相硅胶柱色谱进行分离(100~200目,φ5 cm ×80 cm),干膏与硅胶质量比为1∶30,以石油醚-乙酸乙酯为流动相进行梯度洗脱(95∶5~0∶100),得到6个组分Fr.1(1.759 8 g),Fr.2(3.312 4 g),Fr.3(9.832 1 g),Fr.4(2.985 1 g),Fr.5(1.479 8 g),Fr.6(2.304 2 g).
1.3 色谱条件
1.3.1 制备型HPLC
Agilent ZORBAX SB-C18柱(250mm×21.2 mm,7μm),流动相为甲醇-水,检测波长为280 nm;流速为8mL/min;进样体积3mL,柱温为室温.色谱条件如表1所示.

表1 制备液相色谱条件Table 1 The operation conditions of Pre-HPLC
1.3.2 分析型HPLC
Agilent Lichrosper RP-C18,(5μm,250 mm×4.6mm);流动相为甲醇-水;检测波长为280 nm;流速0.5mL/m in;进样体积 10μL;化合物Ⅰ和化合物Ⅱ进样浓度分别为7.9 mg/mL和5.0 m g/mL;柱温为30℃;化合物Ⅰ和化合物Ⅱ的色谱条件如表2,3所示.

表2 化合物Ⅰ分析液相色谱条件Table2 HPLC operation conditions of compoundⅠ

表3 化合物Ⅱ分析液相色谱条件Table3 HPLC operation conditions of compoundⅡ
1.4 纯品的制备
取Fr.4样品2.985 1 g,用 18 mL甲醇溶解,经离心后取上清液进样,进行高效液相制备色谱分离,按上述制备色谱条件洗脱,收集31~32 m in和34~36 m in两段组分(图 2),减压浓缩,冷冻干燥,得到化合物Ⅰ(0.085 7 g)和化合物Ⅱ(0.962 4 g).
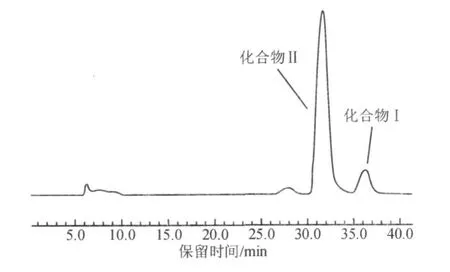
图2 高效制备液相色谱分离Fr.4组分色谱图Fig.2 Preparative chromatogram of Fr.4
2 结 果
2.1 纯度检测
使用高效液相色谱法对分离得到的化合物Ⅰ和化合物Ⅱ的纯度进行检测,经面积归一化法计算,化合物Ⅰ和化合物Ⅱ的纯度均大于98%(图3,4).
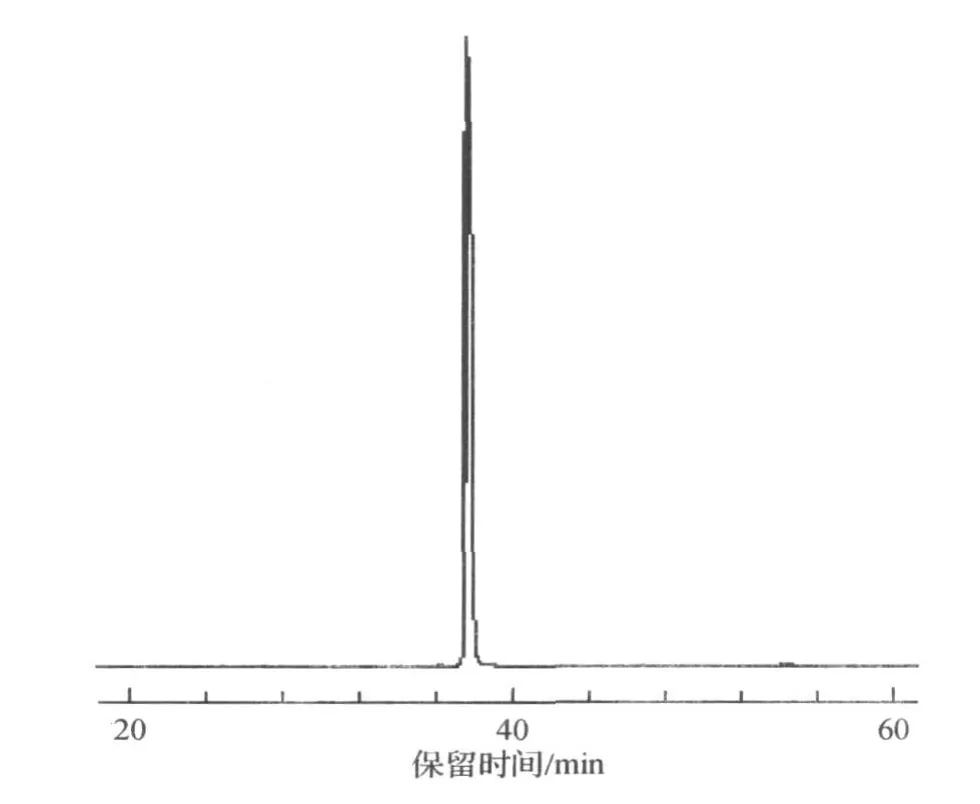
图3 化合物Ⅰ的液相分析图Fig.3 Chromatogram of compoundⅠ
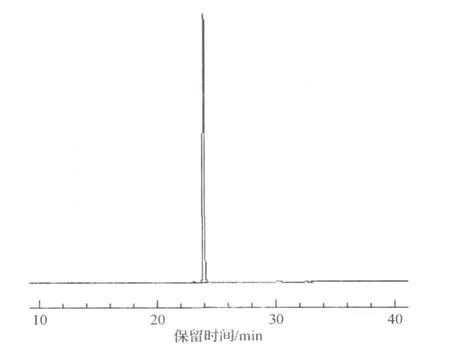
图4 化合物Ⅱ的液相分析图Fig.4 Chromatogram of compoundⅡ
2.2 结构鉴定
2.2.1 化合物Ⅰ的结构鉴定
黄色油状物.1H-NMR(CDCl3,500 MH z,δ)∶0.94(3H,t,J=7.5 Hz,H-11),1.49(2H,tq,J=7.5,7.5 H z,H-10),1.82~1.88,2.03~2.10(2 H,m,H-5),2.36(2H,dt,J=7.5,7.5 Hz,H-9),2.32~2.43,2.61~2.67(2H,m,H-4),3.97(1H,tq,J=6.5,3.5 H z,H-6),4.59(1H,d,J=3.5 H z,H-7),5.30(1H,t,J=8.0 H z,H-8);13C-NMR(CDCl3,125 MHz,δ)∶169.2(C-1),153.8(C-3),148.1(C-3a),125.8(C-7a),114.2(C-8),68.2(C-6),62.3(C-7),27.9(C-9),26.8(C-5),22.1(C-10),19.2(C-4),13.6(C-11).将其核磁数据与文献[10]对照鉴定化合物Ⅰ为洋川芎内酯H.
2.2.2 化合物Ⅱ的结构鉴定
淡黄色油状物.1H-NMR(CDCl3,500 MHz,δ)∶0.95(3H,t,J=7.5 Hz,H-11),1.49(2H,tq,J=7.5,7.5 Hz,H-10),1.86~1.92,2.04~2.07(2H,m,H-5),2.33(2H,dt,J=8.0,7.5 Hz,H-9),2.44~2.56(2H,m,H-4),3.94(1H,ddd,J=8.5,5.5,3.0 Hz,H-6);4.45(1H,d,J=5.5 Hz,H-7),5.29(1H,t,J=8.0Hz,H-8);13C-NMR(CDCl3,125MHz,δ)∶169.2(C-1),153.1(C-3),148.1(C-3a),125.8(C-7a),114.4(C-8),71.7(C-6),67.6(C-7),28.1(C-9),26.5(C-5),22.3(C-10),19.0(C-4),13.8(C-11).将其核磁数据与文献[10]对照鉴定化合物Ⅱ为洋川芎内酯I.
3 结 论
本实验使用D-101型大孔吸附树脂除去川芎提取物中的水溶性杂质后,分别用50%和90%的乙醇洗脱,将川芎提取物粗分成两段,得到大极性段和小极性段,由于洋川芎内酯H和洋川芎内酯I结构中含有两个羟基,极性较大,所以二者主要集中在50%洗脱组分中.然后使用快速正相硅胶柱色谱富集的方法得到洋川芎内酯H和洋川芎内酯I含量较高的组分Fr.4.川芎化学成分复杂,本实验采用大孔吸附树脂进行粗分,有利于后续的分离与纯化.由于洋川芎内酯H和洋川芎内酯I两个化合物的结构非常相似,其液相色谱峰距离很近.使用等度洗脱法对该二者化合物进行分离时,发现分离时间过长、制备效率较低、溶剂消耗较大.因此,本实验使用梯度洗脱法来制备分离洋川芎内酯H和洋川芎内酯I.
本实验采用多波长扫描的方式对洋川芎内酯H和洋川芎内酯I进行制备液相色谱分离,在实验过程中可同时获得320,280,254,210 nm波长下的液相图谱.在这4个波长下Fr.12中所含有的各种化学成分基本都能出峰,并同时呈现于一张图谱中,这样更便于观察各峰之间的分离度,有利于目标成分的收集,从而获得高纯度的化合物单体.
[1]YI Tao,LEUNG K S,LU Guang-hua,et al.Comparative a-nalysis ofLigusticum chuanxiongand related umbelliferous medicinal plants by high performance liquid ch rom atographyelectrospray ionization mass spectrometry[J].Planta Med,2007,73:392-398.
[2]QIHong-yi,SIU SO,CHEN Yan,et al.Senk yunolides reduce hydrogen peroxide-induced oxidative damage in hum anliver H epG2 cells via induction of heme oxygenase-1[J].Chem Biol In teract,2010,183(3):380-389.
[3]YAN Ru,KO N L,LI Song-lin,et al.Pharm acokinetics and metabolism of ligustilide,a major bioactive component in Rhizoma Chuanxiong,in the rat[J].D rug Metab Dispos,2008,36(2):400-408.
[4]洪敏,董自波,朱荃.阿魏酸、洋川芎内酯H和洋川芎内酯1对红细胞的影响[J].时珍国医国药,2003,14(12):738-739.
[5]李绍白,李瀛,李裕林.丁烯基苯酞的改良合成法[J].兰州大学学报,1993,29:256-258.
[6]WANG Pu-shan,GAO Xuan-liang,WANG Yi-xiong,et al.Phthalides from the rhizome ofLigusticum w allichii[J].Phytochem istry,1984,23(9):2033-2038.
[7]KOBA YASHI S,M IMURA Y,NA ITOH T,et al.Chem ical structu re-activity of cnidium rhizom e-derived phthalides for the competence inhibition of p roliferation in primary cultures of mouse aorta sm ooth muscle cells[J].Jpn JPharmacol,1993,63(3):353-359.
[8]石力夫,邓延昭,吴柏生.川芎干燥根茎挥发油化学成分及其稳定性的研究[J].药物分析杂志,1995,15(3):26-30.
[9]LIN Long-ze,HEXian-guo,LIAN Li-zhi,et al.Liquid Ch romatographic-electrospray mass spectrometric study of the phthalides ofAngelica sinensisand chem ical changesof z-ligustilide[J].JCh rom atogr A,1998,810:71-79.
[10]TAKASHI N,TAKAO K,KAZUAK IN,et al.Tw o phthalides fromLigusticum chuanxiong[J].Phytochemistry,1992,31:639-642.
Isolation and purification of senkyunolide H and Iby preparative reversed-phase high performance liquid chrom atography
ZHANG Cui-ping1,LIXing-nuo1,CHEN Yuan-yi1,GE Zhi-wei2,YAN Ji-zhong1
(1.College of Pharmaceutical Science,Zhejiang University of Technology,H angzhou 310032,China;2.Pharmaceu tical Informatics Institute,Zhejiang University,H angzhou 310058,China)
Senkyunolide H and Iwere separated and purified fromLigusticum chuanxiongby using 8 times the amountof80%ethanol to extractLigusticum chuanxiongHort.The ethanolextractwas subjected to D101 macroporous adsorption resin,silica gel,and preparative reversed-phase HPLC for the isolation of target compounds.Their structures were elucidated as senkyunolide H and I by NMR spectral analysis.And thepurity of each compound was above 98%.Thismethod is simple,effective,and reliable for the preparation of senkyunolide H and Isimu ltaneously with good reproducibility and high purity.
preparative reversed-phase high performance liquid chromatography;Ligusticum chuan xiongHort;senkyuno lide H;senkyunolide I
R284.1
A
1006-4303(2011)04-0386-04
2010-03-18
国家重大科技专项“重大新药创制”资助项目(2009ZX09313-036)
张翠萍(1982—),女,河北廊坊人,硕士研究生,研究方向为生物化工,E-mail:zhangcuip314@sohu.com.通信作者:颜继忠教授,E-mail:yjz@zjut.edu.cn.
(
陈石平)
猜你喜欢
中医眼耳鼻喉杂志(2021年2期)2021-07-21 08:53:38
基层中医药(2021年11期)2021-06-05 06:54:38
陶瓷学报(2020年6期)2021-01-26 00:38:14
制造技术与机床(2018年10期)2018-10-13 06:36:56
中成药(2018年7期)2018-08-04 06:04:02
特别健康(2018年2期)2018-06-29 06:13:50
中成药(2017年4期)2017-05-17 06:09:46
核科学与工程(2015年3期)2015-09-26 11:58:24
天然产物研究与开发(2014年6期)2014-04-27 14:15:54
食品科学(2013年6期)2013-03-11 18:20:12
