
心脑联通片质量标准研究
2011-07-29 08:21宋冬梅黎鄂恒李堂秀顾承刚
中国药业 2011年18期
宋冬梅 ,黎鄂恒,吴 宁,李堂秀 ,顾承刚
(1.湖北省荆门市第一人民医院药剂科,湖北 荆门 448000; 2.湖北省中医院药事部,湖北 武汉 430061;3.武汉药谷科技开发有限公司,湖北 武汉 430070)
心脑联通片由灯盏细辛、虎杖、野山楂、柿叶、刺五加、葛根、丹参组方,由心脑联通胶囊[1]改变制剂成型工艺而成,具有活血化瘀、通络止痛的功效,用于瘀血闭阻引起的胸痹、眩晕,症见胸闷、胸痛、心悸、头晕、头痛、耳鸣等,以及冠心病心绞痛、脑动脉硬化、高脂血症见上述证候者。笔者保持原胶囊剂型的提取工艺,仅改变成型工艺,将其制成心脑联通片,并研究了其质量标准,现报道如下。
1 仪器与试药
P3000型高效液相色谱仪(北京创新通恒科技有限公司),包括3000 UV紫外检测器;CQ250型超声波仪(上海超声波仪器厂)。硅胶G(青岛海洋化工厂);羧甲基纤维素钠(国药集团化学试剂有限公司)。心脑联通片(武汉药谷科技开发有限公司制剂室提供,批号为20080301,20080302,20080303,20080304,20080305,20080306);大黄素对照品(批号为110756-200110)、野黄芩苷对照品(批号为110842-200403)、葛根素对照品(批号为 110752-200310),丹参酮ⅡA对照品(批号为110766-200403),均由中国药品生物制品检定所提供,供含量测定用;甲醇为色谱纯,其他试剂均为分析纯。
2 方法与结果
2.1 薄层色谱鉴别
2.1.1 灯盏细辛
取本品10片,除去包衣,研细,称取 2 g,加热水25 mL,研磨5 mim,离心,取上清液,用乙酸乙酯萃取3次,每次20 mL,弃去乙酸乙酯液,水层用稀盐酸调节pH至2~3,用乙酸乙酯萃取2次,每次20 mL,水层备用,合并乙酸乙酯液,水浴蒸干,残渣加甲醇1 mL使溶解,作为供试品溶液。取不含灯盏细辛的自制阴性样品10片,同供试品溶液的制备方法制备,即得阴性对照品溶液。取野黄芩苷对照品适量,加甲醇制成每1 mL含1 mg的对照品溶液。照薄层色谱法,吸取上述3种溶液各2 μL,分别点于同一聚酰胺薄膜上,以冰醋酸为展开剂,展开,取出,晾干,喷以1%三氯化铁乙醇溶液。供试品溶液色谱中,在与对照品溶液色谱相应位置上显相同颜色的斑点,阴性对照品溶液色谱中则无此斑点(见图1 A)。
2.1.2 葛根[2]
取2.1.1项下水液,水浴蒸干,残渣加甲醇适量使溶解,加中性氧化铝7 g,拌匀,挥干,加入50%甲醇50 mL振摇提取,过滤,滤液水浴蒸干,残渣加乙醇1 mL溶解,过滤,作为供试品溶液。取不含葛根的自制样品10片,同供试品溶液的制备方法制备,即得阴性对照品溶液。取葛根素对照品适量,加甲醇制成每1 mL含1 mg的对照品溶液。照薄层色谱法,吸取对照品溶液5 μL、供试品溶液和阴性对照品溶液各10 μL,分别点于同一以羧甲基纤维素钠为黏合剂的硅胶G薄层板上,以氯仿-甲醇-水(7∶2.5∶0.25)为展开剂,展开,取出,晾干,置饱和氨蒸气中放置10 mim,取出,置紫外光灯(365 nm)下检视。供试品溶液色谱中,在与对照品溶液色谱相应位置上显相同颜色的荧光斑点,阴性对照品溶液色谱中则无此斑点(见图1 B)。
2.1.3 丹参
取本品10片,除去包衣,研细,称取3 g,加甲醇30 mL超声处理30 mim,滤过,滤液蒸干,残渣加水30 mL,加热使溶解,放冷,用乙醚提取2次,每次20 mL,合并乙醚液,低温挥干,残渣加甲醇0.5 mL使溶解,作为供试品溶液。取不含丹参的自制阴性样品10片,同供试品溶液的制备方法制备,即得阴性对照品溶液。取丹参酮ⅡA对照品适量,加甲醇制成每1 mL含1 mg的对照品溶液。照薄层色谱法,吸取上述3种溶液各10 μL,分别点于同一以羧甲基纤维素钠为黏合剂的硅胶G薄层板上,以石油醚(30~60℃)-乙酸乙酯(12∶1)为展开剂,展开2次,取出,晾干。供试品溶液色谱中,在与对照品溶液色谱相应位置上显相同颜色的斑点,阴性对照品溶液色谱中则无此斑点(见图1 C)。
2.2 大黄素含量测定
2.2.1 色谱条件与系统适用性试验
色谱柱:Hypersil C18柱(250 mm ×4.6 mm,5μm);流动相:甲醇-0.1%磷酸(80∶20);检测波长:254 nm;柱温:25℃;流速:1 mL/min;进样量 10 μL;检测灵敏度:1.0000 AUFS。理论板数按大黄素峰计算应不低于4000。色谱图见图2。

图1 薄层色谱图
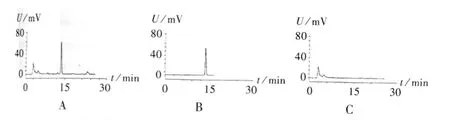
图2 高效液相色谱图
2.2.2 溶液制备
精密称取经五氧化二磷减压干燥24 h的大黄素对照品适量,加甲醇制成每1 mL含30 μg的溶液,即得对照品溶液。取本品5片,除去包衣,研细,称取约0.3 g,精密称定,置具塞锥形瓶中,精密加入三氯甲烷25 mL和2.5 mol/L的硫酸溶液20 mL,密塞,称定质量,置80℃水浴中加热回流2 h,冷却至室温,密塞,再称定质量,用三氯甲烷补足减失的质量,摇匀,分取三氯甲烷液,精密量取10 mL,蒸干,残渣加甲醇使溶解,转移至10 mL量瓶中,加甲醇稀释至刻度,摇匀,0.45μm微孔滤膜滤过,即得供试品溶液,取不含虎杖的自制阴性样品,同供试品溶液制备方法,制备阴性对照品溶液。
2.2.3 方法学考察
线性关系考察:精密称取大黄素对照品0.0093 g,置100 mL量瓶中,用甲醇稀释至刻度,摇匀,分别精密吸取3 mL,置50,25,10,5 mL量瓶中,用甲醇稀释至刻度,配制成质量浓度分别为5.58,11.16,27.9,55.8 μg/mL 的对照品溶液,分别吸取各对照品溶液10 μL,注入色谱仪测定,以大黄素的进样量为横坐标、峰面积为纵坐标绘制标准曲线,得回归方程 Y=4016662 X+37897,r=0.9999(n=5)。结果表明,大黄素进样量在 0.0558 ~ 0.9300 μg范围内与峰面积呈良好的线性关系。
精密度试验:精密吸取同一对照品溶液,按拟订的色谱条件,重复进样6次,测定峰面积。结果的 RSD=0.69%(n=6),表明仪器精密度良好。
稳定性试验:分别精密吸取同一供试品溶液,分别于0,2,4,6,8,10 h时进样,测定峰面积。结果的 RSD为 0.42%(n=6),表明供试品溶液在10 h内保持稳定,能保证测定的时间要求。
重复性试验:取同一批样品6份,按供试品溶液制备方法平行处理并测定,计算含量。结果含量平均值为2.864 mg/g,RSD=1.22%(n=6),表明方法重现性良好。
加样回收试验:取已知含量的样品(批号为20080301,含量为2.864 mg/g)6份,分别精密加入质量浓度为0.4 g/L的大黄素对照品溶液1 mL,依法测定含量,计算回收率。结果见表1。
表1 大黄素加样回收试验结果(n=6)
2.2.4 样品含量测定
取供试品溶液和对照品溶液各10 μL,注入色谱仪,记录峰面积,按外标法计算大黄素的含量,每批样品测定3份。结果见表2。
表2 样品含量测定结果(mg/片)
3 讨论
关于葛根的薄层色谱鉴别,原质量标准中样品处理方法复杂、损失较大。通过试验发现,葛根素水溶性极强,用乙酸乙酯萃取后仍保留在水层,故将提取野黄芩苷后的水层用氧化铝吸附除杂,50%甲醇振摇提取,即可用将葛根素较好地分离出来。原质量标准中使用五元展开剂,配制麻烦,故参照2005年版《中国药典(一部)》葛根药材鉴别项下的展开剂,以三氯甲烷-甲醇-水(7∶2.5∶0.25)为展开剂,效果良好。改进后的鉴别方法操作更简便、灵敏度更高,提高了可操作性和检验的准确性。
关于丹参的薄层色谱鉴别,原质量标准中使用石油醚(60~90℃)作为萃取溶剂,通过试验发现,虽然提取的杂质较少,但丹参酮ⅡA提取效率低,需要较大的点样量方可检出。改用乙醚萃取后,萃取效率提高,正常点样即可检出,且杂质对其无干扰。原质量标准中展开剂为甲苯-乙酸乙酯(18∶1),由于甲苯毒性较大,改用石油醚(30~60℃)-乙酸乙酯(12∶1),展开2次,展开效果良好。改进后的鉴别方法提高了检测的灵敏度,减少了毒性试剂对环境的污染,更加科学环保。
关于虎杖所含大黄素的含量测定,原质量标准中分取氯仿层时较易损失,操作引起的误差较大。将其改为分取氯仿层后精密量取部分氯仿层,通过体积换算得出原液的含量,减小了操作误差;并将流动相由甲醇-0.2%磷酸(75∶25)调整为甲醇-0.1%磷酸(80∶20),增大了甲醇比例,缩短了保留时间,柱温25℃即可获得较好的分离效果,同时降低了酸浓度,减小了流动相对色谱系统的损害,且柱效较高,故选用甲醇-0.1%磷酸(80∶20)作为流动相。改进后的含量测定方法更加简便、高效。
文中建立的定性定量方法简便、快速、重现性好,能有效地控制心脑联通片的质量。
[1]WS-10031-(ZD-0031)-2002,中华人民共和国国家药品监督管理局标准·地标升国标(心系分册)[S].
[2]国家药典委员会.中华人民共和国药典(一部)[M].北京:化学工业出版社,2005:233,52.
猜你喜欢
昆明医科大学学报(2021年8期)2021-08-13
海峡姐妹(2019年8期)2019-09-03
天然产物研究与开发(2019年1期)2019-03-01
中成药(2018年6期)2018-07-11
天然产物研究与开发(2018年1期)2018-02-02
中成药(2017年12期)2018-01-19
中成药(2017年8期)2017-11-22
中成药(2017年3期)2017-05-17
海峡科技与产业(2016年3期)2016-05-17
中国当代医药(2015年8期)2015-03-01
