
烟用香料大柱三烯酮的合成
2011-07-06 08:21陈恩治
食品科学技术学报 2011年3期
陈恩治
大柱三烯酮,即大柱4,6,8-三烯3-酮(megstigma4,6,8-trien-3-one),商品名为烟香酮,化学名为4-(2-亚丁二烯基)-3,5,5-三甲基-2-环己烯-1-酮,分子式为C13H18O,CAS 13215-88-8,是类胡萝卜素化合物经生物降解而成的C13代谢产物之一,于1972年从白肋烟(Burley tobacco)及希腊烟(Greek tobacco)中被分离,并鉴定为天然烟叶中最重要的致香成分之一,其结构式如下:
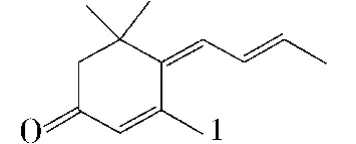
它有4个顺反异构体,如下:
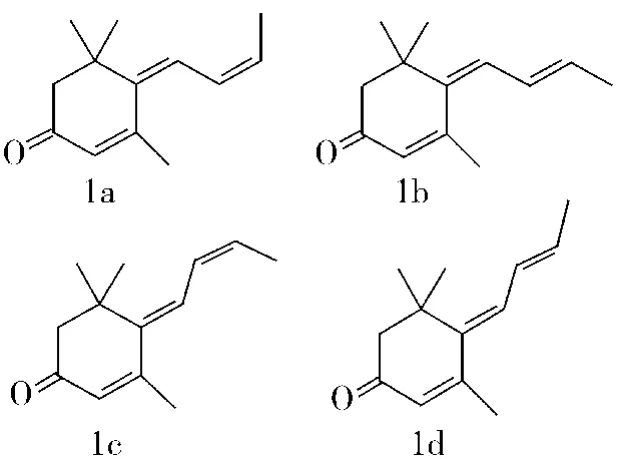
另据报道大柱三烯酮还存在于桂花精油、野葛(kudzu)精油及中国枸杞子(Lycium Chinese M.)果汁中.
大柱三烯酮外观为淡黄色至黄色油状液体,具有强烈而持久的烟草样、甜香、暖香和干香香气,并有辛香的底韵.由于其在天然烟草中含量太少,使一般的烟草味厚度不足,而在卷烟中加入少量的该化合物能明显增强烟香,改善吸味,调和烟气,去除杂气、减少刺激性,因而在烟草香精中使用非常广泛,是调配各种高档烟用香精不可缺少的原料.另外大柱三烯酮还可用于各种日化香精中,也能起到意想不到的效果.在美国FEMA协会公布的GRAS24中,更是将其列入食用香料名录中,FEMA 4663.因此,文献[1]中,Camps甚至预言它将使香精进入一个新的时代.
目前国内外生产销售厂家为数不多,主要有德国Symrise公司及日本长谷川香料公司生产,前者商品名为Tabanon及太白酮,产品含量为50% ~65%(4种异构体混合物),后者为10%的丙二醇溶液,以香基形式销售,由于合成难度大,技术复杂,价格都非常贵.
1 大柱三烯酮的传统合成方法
关于大柱三烯酮合成的文献报道不少,主要有3种方法.
1.1 以紫罗兰酮及其衍生物为原料合成
Rowland[2]将脱氢紫罗兰酮(2)还原成醇(3),经烯丙基重排成大柱4,6,8-三稀-3-醇(4),氧化得到最终产品.这是1965年由美国Reynolds烟草公司提出,并首次实现了大柱三烯酮的人工合成.Trast等同样以脱氢紫罗兰酮(2)为原料,经还原、脱水、亚磺酰化、重排反应得到大柱三烯酮[3].
Aasen等以3-氧代α-紫罗兰醇(5)或3-氧代α-紫罗兰醇乙酸酯(6)为原料,经脱水异构或高温裂解而得到大柱三稀酮[4-7].
1.2 以α-异佛尔酮及其衍生物为原料合成
Takazawa,Tamura以α-异佛尔酮(7)为原料,与格氏试剂甲基溴化镁(8)反应,再与三甲基氯硅烷(9)在六甲基磷酰三胺(HMPT)中反应得到β-异佛尔酮三甲基硅烷烯醇醚(10),在BF3.Et2O催化下与巴豆醛缩合得到3-氧代α-突厥醇(11),后者经甲磺酰氯在1,8-二氮杂环十一烯(DBU)中脱水得到最终产品[8].
Demole等则用酮代异佛尔酮(13)与乙二醇的脱水反应产物为原料,经与各种锂试剂反应,经一系列反应得到大柱三烯酮[9-12].
1.3 以环己酮衍生物为原料合成
Torri,Inskuchi和 Ogawa由 5,5-二甲基 1,3-环己二酮(14)为原料与巴豆醛在二异丙氨基锂(LDA)存在下发生缩合反应得到醇(15),再脱水、用甲基锂甲基化、异构得到大柱三烯酮[13].
通过对以上合成路线的分析研究,不难看出,有些合成路线的原料如脱氢紫罗兰酮、3-氧代-α-紫罗兰醇及其酯或5,5-二甲基1,3-环己二酮国內难以得到,需从头合成相当复杂;有的合成路线反应步骤过长或条件苛刻,有的则使用危险的锂试剂,操作难度大,难以扩大试验等等.
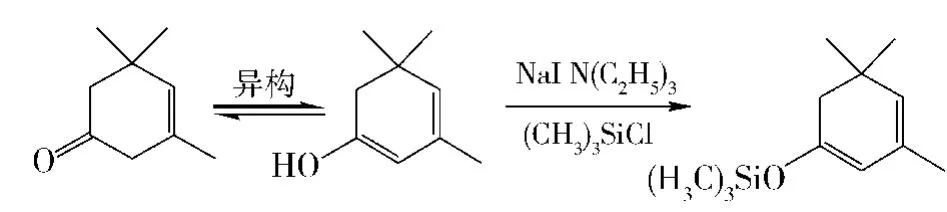
综合考虑,以α-异佛尔酮和巴豆醛为原料的方法,原料宜得,且反应路线比较短,有一定可行性,只是文献[8]报道方法中合成β-异佛尔酮三甲基硅烷烯醇醚以及其与巴豆醛的Aldol反应和产物脱水反应条件较为苛刻,技术难度大,难以工业化.因此合成的关键在于β-异佛尔酮三甲基硅烷烯醇醚的制备,以及寻找适合于工业化的方法,故提出采用α-异佛尔酮为原料,先异构为β-异佛尔酮,再使其在催化剂存在下烯醇化与三甲基氯硅烷直接反应,合成β-异佛尔酮三甲基硅烷烯醇醚,再与巴豆醛反应来合成大柱三烯酮的方法.
反应式见右栏上.
2 实验部分
2.1 试剂与仪器

α-异佛尔酮、甲苯、乙酰丙酮铁(Ⅲ)、二甲基甲酰胺(DMF)、三乙胺、碘化钠、石油醚(60~90℃)、三甲基氯硅烷、巴豆醛、三氟化硼乙醚、甲苯等均为市售化学纯或分析纯试剂,上海国药集团化学试剂有限公司.
CP--3380型气相色谱仪,美国 Varian公司;7890A/5975C型色质联用仪GC--MS,美国安捷伦科技有限公司;色谱柱为SUPELCO PTETM-5(30 m×0.25 mm×0.25μm);DE40型密度仪,瑞士Mettler Toledo公司;WAY--2S型折光仪.
2.2 制备方法
2.2.1 β-异佛尔酮的合成方法
在接有长度为1 m直径为6 cm的精馏柱的2 000 mL三口烧瓶中,加入10 g乙酰丙酮铁(Ⅲ)和1 660 gα-异佛尔酮,升温至沸腾,先全回流半小时,后控制回流比为11∶1,收集沸点为186~196℃馏分,得产品约1 500 g,经GC--MS分析,α-异佛尔酮23%,β-异佛尔酮 75%.
将1 500 g上述α,β-异佛尔酮混合物,装于上述精馏塔的2 000 mL三口烧瓶中,减压精馏,先全回流半小时后,控制回流比3∶1,收集沸点为66~67℃/1.33 kPa的馏分,得990 g产品,β-异佛尔酮含量为99%,得率59%.
2.2.2 β-异佛尔酮三甲基硅烷烯醇醚的合成方法
在装有搅拌、冷凝管、温度计和滴液漏斗的2 000 mL三口烧瓶中,加入400 mL二甲基甲酰胺DMF和320 g(3.2 mol)三乙胺,在搅拌下加入150 g NaI(1 mol)及276 g(2 mol)的β-异佛尔酮,用冰水冷却,t=10℃左右,滴加290 g(2.7 mol)三甲基氯硅烷,温度上升,控制在25℃左右.滴完后在25℃左右搅拌反应8 h,底部有量沉淀析出.过滤,将滤液倒入800 g冰水中,分出油层,水层用250 mL×2石油醚萃取,合并石油醚液与油层,用稀HCl洗涤,再用氯化钠盐水洗至中性,加10 g无水硫酸镁干燥,回收石油醚后,减压蒸馏,收集68~70℃/0.45 kPa的馏分,得321 g产品,含量90%左右,n2D0=1.458 6,d22550.899,产率为76%,MS:210(M 24),195(100),179(45),105(26),75(31),73(69),45(26).
2.2.3 大柱三烯酮的合成方法
在装有搅拌、冷凝管、温度计和滴液漏斗的2 000 mL三口烧瓶中加入上述321 g(90%,1.37 mol)β-异佛尔酮三甲基硅烷烯醇醚,500 mL无水甲苯和115 g(99%,1.64 mol)巴豆醛,冷却至-25℃,开始滴加 BF3·Et2O194 g(1.37 mol),温度有所上升,约1 h滴完,控制反应温度在-20℃左右,滴完后再在-20℃左右反应8 h.然后由滴液漏斗慢慢加入由126 g Na2CO3和600 g水配成的溶液,滴完后搅拌10分钟,倒入2 000 mL分液漏斗中,分去水层,油层用水洗至中性,回收甲苯后,经减压蒸馏,收集115~120℃/0.27 kPa的馏分,得120 g产品,含量为75%左右,n2D0=1.572 0,d22550.990,含异构体1a约8%、1b约37%、1c约6%、1d约25%,产率为46.1%,MS:190(M 100),175(77),148(77),147(83),133(77),122(55),119(63),105(63),91(77),69(63),55(52),41(77).如以β-异佛尔酮来计,两步反应总得率为35%(文献[8]报道,以α-异佛尔酮来计为40%).
3 结果与讨论
3.1 β-异佛尔酮三甲基硅烷烯醇醚的合成
1)实验中参照文献[14]的方法,先将异佛尔酮在乙酰丙酮铁(Ⅲ)催化下异构为β-异佛尔酮,经分馏得到含量为99%的β-异佛尔酮,得率59%,分馏剩余的α-异佛尔酮可以再套用异构分馏.
2)β-异佛尔酮在碘化钠催化和缚酸剂三乙胺存在下与三甲基氯硅烷于二甲基甲酰胺DMF中反应,来制备β-异佛尔酮烯醇硅醚.反应中β-异佛尔酮先异构为烯醇式,再与硅烷发生醚化反应.
3)该方法避免已有文献[8]中报道的采用格氏试剂甲基溴化镁CH3MgBr(需使用剧毒的溴甲烷来制备)来异构,并且用二甲基甲酰胺DMF代替剧毒的六甲基磷酸三酰胺HMPT溶剂,另外使β-异佛尔酮与三甲基氯硅烷反应在常温进行,避免文献报道采用的-20℃低温,缩短了反应时间,而得率相差无几.
4)实验中发现碘化钠的量对反应时间影响比较大,越多则反应越快,主要是碘化钠先与三甲基氯硅烷进行卤素置换反应,生成活性更高的三甲基碘硅烷,从而加快反应进程.

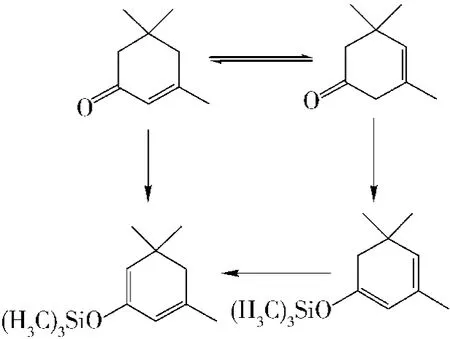
5)实验中发现,在β-异佛尔酮与三甲基氯硅烷反应过程中,有部分β-异佛尔酮异构为α-异佛尔酮,生成α-异构体硅醚;而用稀盐酸洗料时的温度对产品影响较大,温度过高,β-异佛尔酮烯醇硅醚异构为α-异构体硅醚,且容易水解为原料α-异佛尔酮.
因为从分子结构来看,α-异佛尔酮远比β-异佛尔酮稳定,因其具有共轭效应,同样生成的硅醚衍生物也是α-异佛尔酮比β-异佛尔酮稳定,而此α-异构体硅醚同样能与巴豆醛反应,生成的却是副产物.
3.2 大柱三烯酮的合成
1)文献[8]报道利用β-异佛尔酮三甲基硅烷烯醇醚合成大柱三烯酮中,先得到3-氧代-α-突厥醇.而最后一步中先要在DBU与甲基磺酰氯反应得到甲基磺酸酯,然后在甲苯中脱甲基磺酸得到大柱三烯酮,反应步骤长,操作繁琐.
2)实验中用无水甲苯替代了文献[8]报道采用的毒性较大、挥发度较高的二氯甲烷,反应效果相差不大.
3)实验中发现硅醚与巴豆醛在三氟化硼乙醚的催化下发生加成反应后,蒸馏即得到了大柱三烯酮,经色谱跟踪发现反应结束并洗涤后,并没有大柱三烯酮的峰出现,而是在蒸馏回收溶剂过程中受热直接脱水所致.这主要是由于中间产物3-氧代-α-突厥醇中的羟基受结构影响,受热非常容易脱水而形成共轭双键,从而转变为产品大柱三烯酮.而实验中采用高沸点甲苯替代低沸点的二氯甲烷,恰恰使中间体3-氧代-α-突厥醇在受热回收溶剂过程中由于温度更高而更容易进行,完全脱水成为产品.
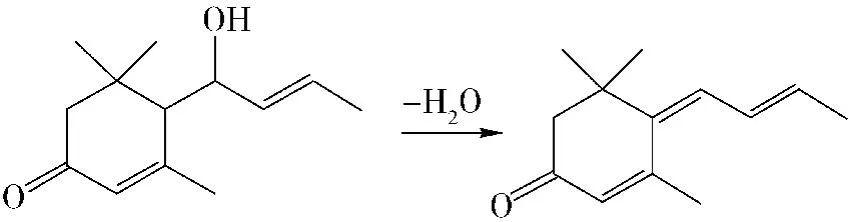
4)文献[8]报道,反应温度最好为-40~-60℃,而实验中发现,反应温度为-5~-10℃与反应温度为-40℃,所得到的实验结果差不多.
5)实验中曾采用精密分馏来提纯,尽管可以得到含量≥90%,但损失非常大,主要是产品分子结构中有很多共轭双键,特别容易聚合,蒸馏的脚子非常多.
4 结 论
1)以α-异佛尔酮为原料,经乙酰丙酮铁(Ⅲ)异构得到β-异佛尔酮,再与三甲基氯硅烷在碘化钠催化和缚酸剂三乙胺存在下于DMF中反应,得到β-异佛尔酮三甲基硅烷烯醇醚,得率45%.
2)β-异佛尔酮三甲基硅烷烯醇醚与巴豆醛经Aldol反应,产物经蒸馏脱水一步合成了大柱三烯酮,得率46%.
3)该方法原料易得,操作相对简便,避免了文献[8]报道的采用剧毒原料、超低温反应等一系列不利因素,比较适合于大规模的生产.
[1] 黄致喜,王慧辰.萜类香料化学[M].北京:中国轻工业出版社,1999:210.
[2] Rowland R L.Process for preparing 4-(2-butenylidene)-3,5,5-trimethyl-2-cyclohe xene-1-one:US,3268589[P].1966--08--23.
[3] Trast B M,Stanton J L.New synthesis methods 1,3-Alkylative carbonyl transposition[J].J.Am.Chem.Soc,1975,97:4018.
[4] Aasen A J.Tobacco Chemistry.15.new tobacco constituents.The structures of five isomeric megastigmatrienones[J].Acta.Chem.Scand,1972,26(6):2573--2576.
[5] Brunke ernst-joachim.Process for the preparation of 4,4,7-trimethyl-3,4,7,8-tetrahydro-2(6H)-naphthaleneone:US,4753924[P].1988--6--28.
[6] 李俊,蒋思翠.一种巨豆三烯酮香料的合成方法:中国,1434017[P].2003--08--06.
[7] 谢冰,孔宁川.一种巨豆三烯酮的合成方法:中国,1803749[P].2006--07--19.
[8] Takazawa O,Tamura H.New synthesis of megastigma-4,6,8-trien-3-ones,3-Hydroxy-j8-ionol,3-Hydroxy-£-ionone,5,6-Epoxy-3-hydroxy-£-ionol,and 3-Oxo-a-ionol[J].Bull.Chem.Soc.Japan,1982,5:1907.
[9] Demole E,Engyist P.Novel synthesis of 3,5,5-trimethyl-4-(2-butenylidene)-cyclo hex-2-en-l-one,a major constituent of burley tobacco flavour[J].Helv.Chim.Acta,1974,57:2087.
[10] Daniel K,Frank K,Erich W.Process for the preparation of a cyclic unsaturated ketone:EP,356869[P],1990--03--07.
[11] 王建林,杨少龙,许炎妹,等.巨豆三烯酮的合成及表征[J].光谱学与光谱分析,2005,25(3):467--469.
[12] 伊藤信彦,木之下公男,江藤武显.制备4-烷叉基-3,5,5-三甲基-2-环己烯-1-酮:JP,93097763[P].1993--04--40.
[13] Torri S,Inskuchi T,Ogawa H.A synthesisof 4-(trans-2-butenyliene)-3,5,5-trimethyl-2-cyclohexen-1-one[J].Bull.Chem.Soc.Japan,1979,52:1233.
[14] Bellut hans.Process for the preparation of beta-isophorone from alpha-isophorone:US,4845303[P].1989 04--27.
猜你喜欢
食品安全导刊(2021年21期)2021-08-30
生物化工(2021年3期)2021-07-10
传奇·传记文学选刊(2019年7期)2019-07-19
故事会(2019年7期)2019-04-22
猪业科学(2018年4期)2018-05-19
婚姻与家庭·性情读本(2017年3期)2017-04-27
故事会(2016年10期)2016-05-21
河北医科大学学报(2011年11期)2011-03-25
民间文学(2009年10期)2009-10-30
