
我国羊种3型布鲁氏菌的多位点序列分型研究*
2011-06-06 13:24周晓艳陈燕芬崔步云陈泽良赵鸿雁朴东日李兰玉王金桃
中国人兽共患病学报 2011年5期
周晓艳,陈燕芬,崔步云,陈泽良,姜 海,赵鸿雁,朴东日,李兰玉,王金桃
2.中国疾病预防控制中心,传染病预防控制所布鲁氏菌病室,北京 102206;
3.军事医学科学院疾病预防控制所,北京 100071
多位点序列分型(Multiple locus sequence typing,MLST)是一种通过对多个(一般为7个)管家基因的测序,用核苷酸序列变异来发现细菌型别差异的分型方法[1]。该方法在实验过程的可操作性及实验结果的可靠性之间取得了平衡[2]。由于其结果明确,在不同的实验室之间具有良好的可比性[3],因此被广泛的应用于细菌的分型研究,并且对于某些菌株具有良好的分辨能力[4]。
布鲁氏菌病(brucellosis,布病)是由布鲁氏菌(brucella,布氏菌)属的细菌侵入机体,引起的传染—变态反应性的人兽共患病。近年来,我国人间布病的发病数逐年递增,已远远超过历史最高水平。病原学监测研究显示,近年在我国引起人发病的布氏菌主要是羊种3型,为了进一步了解该型布氏菌的遗传特征,以及与其它型之间的关系,我们选取了分离自我国不同省份的47株羊3型布氏菌,进行了MLST分型,研究其遗传多态性。将其ST型与国外菌株的ST型进行比较,从而了解与国外菌株间的遗传进化关系,尝试解释近年愈演愈烈的国内布病疫情。
1 材料与方法
1.1 布鲁氏菌菌株 收集2004-2009年分离自我国的47株羊种3型布氏菌。菌株由中国疾病预防控制中心传染病预防控制所布鲁氏菌病室分离、收集、鉴定和保存。
1.2 细菌培养与基因组DNA的提取 从菌种库中取出冻干布氏菌株,无菌操作打开菌种管,加入布鲁氏菌肉汤0.5mL使冻干物溶解。取0.1mL溶解物均匀涂布于布鲁氏菌琼脂培养基上,同时接种于布鲁氏菌肉汤培养基,置37℃培养48h。经纯培养、血清凝集、噬菌体裂解、三胜黄素凝集鉴定复核后收集菌体,用热灭活的方式灭活细菌,然后用基因组提取试剂盒(天根生物,北京)提取基因组DNA,基因组的提取按试剂盒说明书进行。提取的基因组DNA于-20℃保存备用。
1.3 PCR扩增 参考文献选择7个管家基因(dnaK 、gyrB 、trpE、aroA 、cobQ 、gap、glk)、1 个外膜蛋白基因(omp25)和 1个基因间区int-hyp作为MLST的靶标基因[5],并合成相应的引物。选取的靶标基因、引物、序列及扩增长度如表1所示。PCR的反应体系(50μL):DNA1μL、引物(10μmol/L)2μL、TaqDNA 聚合酶(2.5U/μL)0.25μL、10×Taqbuffer(Mg2+1.5mol/L)5μL 、dNTPs(2.5 mmol/L)4μL、去离子水:38μL。PCR反应条件为:95℃预变性 5min;94℃变性30s,63℃退火30s,72延伸1min,30个循环;72℃延伸10min。

表1 MLST靶标基因、引物序列与扩增产物大小Table1 Oligonucleotidesequencesusedfortheamplificationandsequencingofninegeneticloci
1.4 PCR产物检测与测序 PCR产物用1%琼脂糖凝胶电泳分析,检测扩增是否特异或成功,扩增特异产物直接用于测序。PCR产物纯化后,用扩增引物进行双向测序,测序由擎科测序公司进行。
1.5 序列分析 整理每株菌各个基因的测序结果,编辑序列,使序列长度范围与文献中给出的序列一致。将测定序列分别与相应基因的等位基因型的序列进行比较,明确序列是否与某个等位基因型的序列一致,如果一致,则定义为某个等位基因型,否则定义为一个新的等位基因型。利用MLST在线工具(http://pubmlst.org/perl/mlstanalyse/mlstanalyse.=pubmlst)NRDB、SplitsTree、LIAN、BURST、Treedrawing等工具,对序列的等位基因型、序列型分离、连锁平衡、聚类、进化树等进行分析,确定序列型、探讨菌株间的进化关系。
2 结 果
2.1 ST28型的鉴定 47株菌中的28株与已知的ST8型的各等位基因型是一致。另外 19株菌的omp25基因与已有的等位基因型不同,被定义为1个新的等位基因型,即ST28,见表2。
表显示ST28与ST8有1个位点(omp25)的差异,其中ST8的omp25等位基因型为8型,而ST28的omp25等位基因型不属于国外发现的已知的10个等位基因型,本研究我们将其定义为等位基因型11,而ST型也在原有的27型的基础上,命名为ST28型。
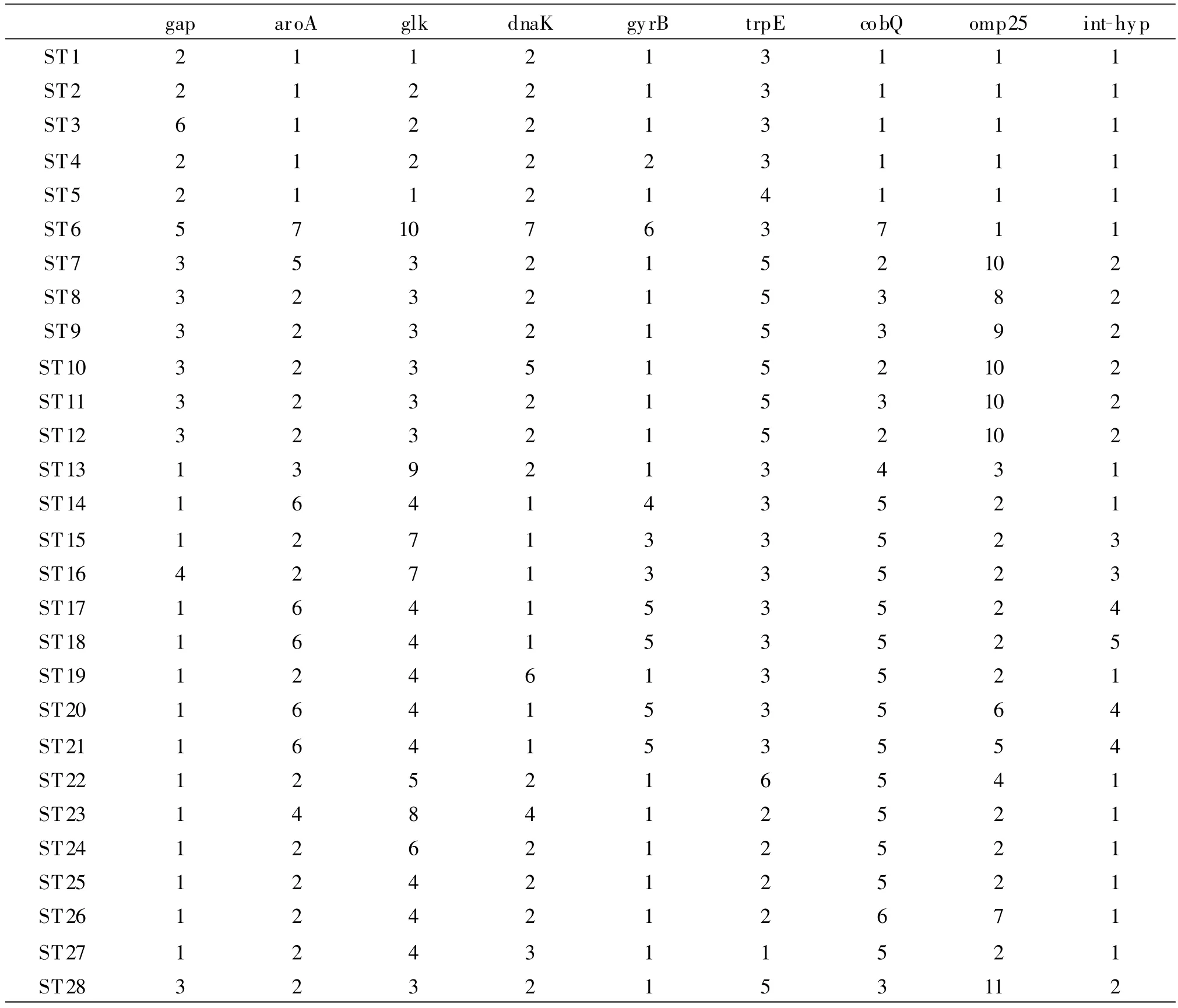
表2 28个ST型的各等位基因谱Table2 28ST-typeallelesspectrum
2.2 47株菌的ST型分型地域分布 本研究中47株菌经经典方法鉴定为布氏菌属羊种3型。ST分型数据分析显示,其中19株分离自山西、陕西、广东、河北、湖南的菌株,其omp25基因与已知的等位基因型不同,为等位基因11型,ST分型为ST28型,它们均为2008、2009年新分离到的菌株;分离自辽宁的28株菌所有的等位基因型与ST8的各等位基因型一致,表明这些菌株属于ST8型,它们分离自 2004-2008 年,分别为 11、4、3、7、3 株。
2.3 不同基因位点的多态性 用LIAN连锁分析工具,对ST型的等位基因型进行了连锁不平衡分析显示,VD为4.8308,VE为 1.8175,SIA为 0.2072。9个位点的平均遗传多态性(H)为 0.6999+/-0.0318。9个位点即 Gap、aroA 、glk、dnaK、gyrB、trpE、cobQ、omp25和int-hyp的遗传多态性分别为0.6878、0.7063、0.836、0.627、0.5291 、0.6878、0.7434、0.8254和 0.6561。
遗传多态性反映不同基因位点的多态性变异程度,数值介于0~1,数值愈大,其变异度越大。各位点多态性分析显示,glk和omp25的变异度最大,这两个位点的等位基因型均为10,远远高于其它几个位点,因此,菌株的多态性将主要来自于这两个位点。我们本次鉴定的新ST28型,就是来自omp25的差异。
2.4 对ST28采取的不同的分析方法 为了进一步分析ST28型的特点,对所有已鉴定的ST型进行了分离分析。利用Splits tree工具,对ST型的数据进行分裂分解分析(Split decomposition analysis),从图1中可以看出,ST28与其它几个均属于羊种布氏菌的 ST型聚在一起,与 ST8、ST9、ST11高度同源,只有1个等位基因om p25的差异。它们均由ST12分离而来。与其它种的布氏菌相比,羊种菌的这几个ST型是聚集在一起的,说明羊种布鲁氏菌在基因水平上是高度保守的。
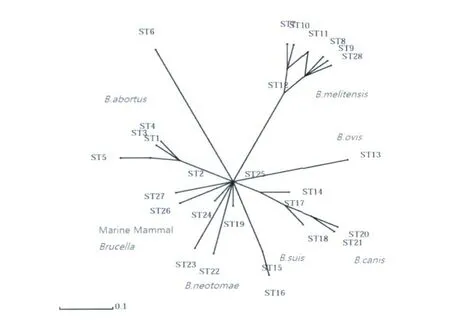
图1 不同ST型间的Splits TreeFig.1 Splits Treeofdifferent ST-type
对不同ST型间的进化关系采用neighbor joinning计算方法进行分析,各个ST型间的进化关系如图2所示。羊种布氏菌的几个ST型处于同一个进化分支上,且高度集中,说明羊种布氏菌是一种高度进化,且变异度小的一种布氏菌。ST28型与ST8、ST9遗传关系很近,处于同一个进化层次。
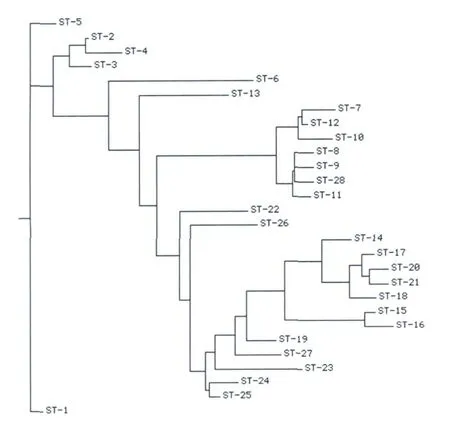
图2 不同ST型间的进化关系Fig.2 Differentevolutionary relationships between ST-type
BURST(Based U pon Related Sequence Type)是一种基于相关序列型的分型方法,用于根据细菌的等位基因型谱将细菌分为不同的群,计算每个序列型的单个位点变异数 SLV(Single Locus Variants)、双位点变异数(DLV)和卫星数,鉴定可能的祖先序列型等。利用BURST分析28个ST型。布氏菌的28个ST型之间,除了少数的几个有联系之外,更多的ST型是散在存在的。这一方面提示,布氏菌的ST型太少,难分析出不同型间的变异关系,另一方面也表明,布氏菌是一个高度保守的细菌,ST型变异小。
3 讨 论
布氏菌分子流行病学研究,对了解布氏菌的菌株遗传变迁,流行株的形成等具有重要意义。多位点序列分型(multilocus sequence typing,MLST)作为一种准确的分子分型方法于1998年问世以来[6],以其高分辨率[7],可重复性好,可积累等[8]优势而得到广泛关注。随着测序速度的提高,数据库积累快速增长,在病源微生物分类方面的优势突显。通过对多个管家基因的测序结果,用核苷酸序列变异来发现细菌型别差异的分型方法[9],广泛用于研究细菌的流行病学、群体生物学、致病性和进化[10],成为分子流行病学研究新方法。
布氏菌是一个基因组较为保守的细菌,国内外对分型作过很多研究。崔步云等用REP-PCR、PFGE以及脂肪酸分型等方法作种属分类[11-13],姜海等用MLVA将31株羊3型布氏菌分为了9个基因型[14]。而将MLST分型用于中国分离的布鲁氏菌尚未见报道。与其它细菌的ST型相比,布氏菌的ST型相对较少,Ad rian[5]等分析了160株分离自英国、美国、爱尔兰、法国、新西兰等(不包括中国)约30个国家的160株菌,共发现了27个ST型。我们以此对2004至2008年分离自中国辽宁、山西、陕西、河北、广州、湖南菌株为材料进行检测,发现辽宁菌株与国外的ST8羊种菌一致;而其余省份菌株为1个新的ST型,我们命名为ST28,该型主要是来自om p25的碱基变化,该型国内外均未见报道。
近年来,我国流行的布氏菌主要是羊种和牛种菌,患布病羊是我国人间布病最主要的传染源,近年致病菌主要是羊种3型布氏菌。近5年我室从患者体液分离获得357株布氏菌,羊种菌占99%,羊种3型有277株。一般羊种3型对人、畜均有较强的毒力和侵袭力,且致病力也较强,易引起布病的暴发和流行,且疫情重。患者主要临床表现为长期发热、出汗、乏力和关节疼痛等[15]。布病是自然疫源性的疾病,本次研究结果中,2004-2008年分离自辽宁的28株均为ST8,国外分离菌株也有ST8型,分析可能是当地作为疫源地保留和在当地流行引起发病的菌株。而山西、陕西、广州、湖南、河北 5省 2008、2009年分离的菌株为同一型别,即新发现的ST28,分析原因为近年国内传染源流动,引起国内布病流行的主要菌株,它们可能是我国布病的主要传染源。
本研究中,我们选取的菌株数量有限,仅涉及近年主要引起人感染的部分菌株;对我国流行情况的分析具有一定的局限性。在后续的研究中,我们将选取更多种型、不同年代和地区的菌株进行更为全面的研究,并完善中国布氏菌株MLST数据库。
[1]H anage W P,Feil EJ,Brueggem enn AB,et al.Multilocus sequence typing:strain characterization,popu lation biology,and patternsof evolutionary descent.In Persing,DH,et al.(eds),Molecu lar Microbiology:diagnositic principles and practice.Am ercan Society Press[M].Washington DC,235-243.
[2]Maiden MC J,Bygraves JA,Feil E,et a1.Multilocus sequence typing:a portable approach to the identification of clones within populations of pathogenic microorganisms[J].Proceedings of the NationalAcademy of Science,1998,95:3140-3145.
[3]Katetishvili M,Stne O C,Chen Y,et a1.Mu ltilocus sequence typing hasbetter discriminatory ability for typing Vibrio cholerae than does pulsed-field electrophoresis and provides ameasure of phylogenetic relatedness[J].J C lin Microbiol,2003,41(5):2191-2196.
[4]Helgason E,Tourasse N J,Meidal R,et al.Multilocus sequence typing scheme for bacteria of bacilluscereus group[J].Appl Environ Microbiol,2004,70(I):191-201.
[5]Ad rian MWhatmore,Lorraine L Perrett and A lastair PMacMillan:Characterisation of the genetic diversity of Brucella by multilocus sequencing[J].BMCMicrobiology,2007,10 1186/1471.
[6]Maidenm CJ,Bygraves JA,Feil E,et al.Multilocus sequence typing:a portable approach to the identification of clones with in populations of pathogenic microorganisms[J].Proceedingsof the National Academy of Science,1998,95:3140-3145.
[7]Brian G S,Martin C J.Bacterial population genetics,evolu tion and epidem iology[J].Philosoph ica l Transactions of the Royal Society B,1999,354:701-710.
[8]Gogarten JP,DoolittleW F,Jeffrey G L.Prokaryotic evolu tion in light of gene transfer[J].Molecular biology and evo lution,2002,19:2226-2238.
[9]H anage WP,Feil EJ,Brueggemenn AB,et al.Multilocus sequence typing:strain characterization,population biology,and patterns of evolu tionary descen t.In Persing,DH,et al.(ed s),Molecular Microbiology:diagnosis principles and practice.Amercan Society Press.Washington DC,pp.235-243.
[10]Aanensen DM,Spratt BG.Themu ltilocussequen cetyping network:mlst.net.Nucleic Acids Research,2005,33:728-733.
[12]崔步云,尹继明,李兰玉.布鲁氏菌的 Rep-PCR分型研究[J].疾病监测,2005,20(8):397-340.
[13]崔步云.布鲁氏菌脉冲场凝胶电泳分型研究[D].山西医科大学硕士论文,2006.
[14]ZHAO ZX,CUIBY,LILY,et al.Analysisof Brucella Cellular Fatty Acid s Frontiers of medicine in China[J].F ron tiers of Medicine of China.2010,4(2):216-219.
[15]Jiang H,Mao LL,Zhao HY,et al.MLVA typing and antibiotic susceptibility characterization of Brucella Isolates from human in Liaoning,China[J].Transactions of the Royal Society of Tropical Medicine and H ygiene,2010,104(12):796-800.
[16]肖东楼,冮森林,崔步云,等.布鲁氏菌病防治手册[M].北京:人民卫生出版社,2008:38-52.
猜你喜欢
传染病信息(2022年6期)2023-01-12
川北医学院学报(2022年6期)2022-06-24
智慧健康(2021年17期)2021-07-30
现代畜牧科技(2021年4期)2021-07-21
智慧农业导刊(2021年20期)2021-03-09
中国产前诊断杂志(电子版)(2020年1期)2020-05-21
遵义医科大学学报(2020年6期)2020-02-05
中国医药指南(2017年30期)2017-01-15
中国民族医药杂志(2016年9期)2016-05-09
浙江农业科学(2016年11期)2016-05-04
