
药食两用类食品中赭曲霉毒素A的高效液相色谱-荧光检测方法
2011-04-06 09:40傅武胜邱文倩郑奎城吕华东严小波
食品科学 2011年14期
傅武胜,邱文倩,郑奎城,吕华东,郭 斌,严小波
(1.福建省疾病预防控制中心,福建 福州 350001;2.福建医科大学 福建省疾病预防控制中心教学基地,福建 福州350001;3.福建中医药大学,福建 福州 350001;4.福州大学化学化工学院,福建 福州 350001)
药食两用类食品中赭曲霉毒素A的高效液相色谱-荧光检测方法
傅武胜1,2,邱文倩1,郑奎城1,吕华东1,郭 斌3,严小波4
(1.福建省疾病预防控制中心,福建 福州 350001;2.福建医科大学 福建省疾病预防控制中心教学基地,福建 福州350001;3.福建中医药大学,福建 福州 350001;4.福州大学化学化工学院,福建 福州 350001)
目的:采用免疫亲和净化和高效液相色谱技术,建立淀粉、糖类药食两用食品中赭曲霉毒素A的测定方法。方法:样品粉末经甲醇-水(8∶2,V/V)涡旋、超声及振摇提取,提取液以磷酸盐缓冲液稀释后,用商品免疫亲和柱净化,含有赭曲霉毒素A的甲醇洗脱液用高效液相色谱技术分析,C18反相色谱柱分离,荧光检测器测定。结果:赭曲霉毒素A的最低检出浓度为1.0μg/kg(RSN=3);在0.5~100ng/mL范围内,峰面积与质量浓度呈线性关系(r=0.9995);以不含赭曲霉毒素A的太子参、莲子、薏苡、麦冬和龙眼肉为加标基质,加标水平为1~8μg/kg时,平均回收率在81.8%~107%之间,RSD为1.66%~15.0%(n=3)。结论:免疫亲和柱能和高效液相色谱-荧光检测相结合取得较为满意的结果,准确度高、精密度好,满足欧盟对食品饲料中OTA检测方法的要求,适合于淀粉、糖类药食两用食品中赭曲霉毒素A的测定。
药食两用食品;赭曲霉毒素A;免疫亲和净化;高效液相色谱法
赭曲霉毒素(ochratoxins)是曲霉属和青霉属的某些菌种产生的一种具有致癌、致畸、免疫抑制和肝肾毒性的有机酸类真菌毒素,有A、B、C、D四种化合物[1],国际上较为关注的是赭曲霉毒素A(OTA),其在霉变谷物、饲料中较为常见。WHO/FAO食品添加剂和污染物联合专家委员会制定了OTA的暂定每周最大耐受剂量[2],欧盟评估了人群膳食暴露水平[3]。国际食品法典已制定了食品中OTA的限量和良好操作规范[2],以降低OTA的健康危害。我国规定谷类和豆类OTA的限量为5μg/kg[4],欧盟对OTA的控制更为严格、具体[3],2010年欧盟增加了香辛调味料和甘草中OTA的限量,分别不能超过15~30μg/kg和20μg/kg[5]。甘草、橙花、咖啡、茶藨子、酸橙花、黑胡椒、胡荽、姜黄、干姜和人参等植物和/或芳香调味料OTA的污染较为普遍[6-10],有些,如甘草污染水平较高,50%的样品含量超过5.3μg/kg[9-10]。含甘草提取物的食品也检出0.34~2.96μg/kg的OTA,儿童每周由此摄入的OTA相当于PTWI的8.94%[11]。
香辛调味料、植物药中OTA的检测方法有薄层色谱法[12]、酶联免疫吸附法[8,13]、荧光光度法[14]、高效液相色谱法[10,15-17]和液相色谱/质谱法[10,14]等。薄层色谱法法为半定量方法,灵敏度较低,重复性差,如未经过充分净化,杂质干扰比较严重。ELISA法被用于初筛,成本低,但用于检测中样品时假阳性率较高。液相色谱/质谱法是一种确证性方法,检出限低至0.3μg/kg,但仪器也十分昂贵,分析成本高。高效液相色谱法结合荧光检测器,灵敏度较高,定量结果准确。生物样品中OTA含量低(μg/kg级),基体复杂,干扰较多,因此IAC(immunoaffinity column)净化效果更好。虽有免疫亲合柱上显色快速筛查法[18]用于色素含量较高的甘草、姜、肉豆蔻、黑胡椒、白胡椒和辣椒,以及IAC-TLC法[12]用于测定绿咖啡中OTA测定的报道,但应用最普遍、效果最好的还是IAC-HPLC法,已经有较为成熟的商品IAC柱出售,国外有用于植物药的报道[10,17]。此外,也有商品多功能亲和柱,可用于同时净化、检测人参、干姜和松果菊中的OTA和黄曲霉毒素[7,16]。国内应用该法于香辛料和藤蔓水果干中OTA的检测[14],但未见用于药食两用类食品样品。因此本实验从福建省主产的淀粉或糖含量较高的6种药食两用食品入手,发展基于IAC净化的HPLC-荧光检测器检测技术,为开展污染调查提供良好的技术手段。
1 材料与方法
1.1 材料与试剂
太子参、莲子、薏苡、泽泻、麦冬、龙眼干样品在2009年6月至2010年3月间,购于福州市各大连锁药店、超市和福建省部分产地,样品经福建中医药大学药学院吴锦忠教授鉴定为太子参(Pseudostellaria heterophylla (M iq.) Pax ex Pax et Hoffm)、泽泻(Alisma orientale (Sam.) Juz.[A.plantago-aquatica L.var. orientale Sam.])、莲子(Nelumbo Nucifera Gaertn.)、薏苡(Coiχ lacroyma-jobi L. var. ma-yuen (Roman.) Stapf)、麦冬(Ophiopogonjaponicus (Thunb.) Ker-Gawl.)、龙眼肉(Euphoria longan (Lour.) Steud.)。样品处理方法:用高速粉碎机粉碎1~2min,每份样品粉碎完后,去除参与粉末,再用75%乙醇清洗和消毒粉碎机腔体和机盖;用电吹风吹干表面后,再粉碎下一个样品;样品粉末冷却至室温后,装入密封袋中于冰箱冷藏。
免疫亲和柱为德国LC Tech公司的OtaCLEANTM(3mL/支)和美国Vicam公司的Afla TestR-P(1mL/支)、50mL聚丙烯塑料离心管(美国Corning公司)、0.45μm水系微孔滤膜(直径50mm)、玻璃微纤维滤纸(直径110mm)。
甲醇(色谱纯) 美国Fisher公司;冰乙酸、乙醇(均为优级纯);氯化钠、磷酸氢二钠和磷酸二氢钾(均为分析纯);赭曲霉毒素A标准液(OTA,50μg/mL) 美国Supelco公司。
OTA标准储备液(10μg/mL)的配制:移取OTA标准液(50μg/mL),移入10mL棕色容量瓶中,用甲醇定容,涡旋混匀,-20℃放置备用,进一步配制为100ng/mL的标准使用液。磷酸盐缓冲液(PBS,pH7.2)的配制:称取10.03g Na2HPO4溶于140mL超纯水中,再称取1.932g KH2PO4溶于70mL超纯水中,将两种溶液混合后加入8.5g NaCl,充分溶解混匀,得到母液,母液稀释5倍后供使用。
1.2 仪器与设备
LC-20AD型高效液相色谱仪(配RF-10AXL荧光检测器) 日本Shimadzu公司;DFY-80型摇摆式高速万能粉碎机 温岭市林大机械有限公司;CP225D型电子天平(感量0.1mg) 瑞士Sartorius公司;YP202N型电子天平(感量0.01g) 上海精密科学仪器有限公司;G-560E漩涡混合器 美国Scientific Industries公司;18型高速离心机、Allegra 25R型台式高速冷冻离心机 美国Beckman公司;溶剂抽滤装置 河北津腾公司;KQ-500B型、KQ3200型超声波清洗器 昆山市超声仪器有限公司;固相萃取装置 美国Phenomenex公司;振荡器 美国IKA公司;精密移液枪(100、200、1000μL)美国Gilson公司;5mL移液枪 上海大龙公司;超纯水机 北京历元电子仪器公司。
1.3 标准曲线的建立
1.3.1 标准曲线绘制
准确移取适量OTA标准使用液(100ng/mL),加入甲醇,涡旋30s,配成质量浓度为0.500、1.250、2.500、5.000、10.000ng/mL的标准系列,进行HPLC分析。以峰面积(Y)对OTA的浓度(X,ng/mL)以最小二乘法作线性回归,得到标准曲线。
1.3.2 液相色谱分析条件
采用自动进样方式,将适宜浓度的OTA溶液注入高效液相色谱仪。色谱条件如下:色谱柱:Shiseido C18柱(4.6mm×150mm,3μm);柱温:35℃;流动相:甲醇-2%冰乙酸(65∶35,V/V);流速:0.8mL/min;荧光检测器:激发波长(λex)为333nm,发射波长为(λem) 447nm;进样量:20μL。
1.4 样品制备和OTA测定
1.4.1 样品的提取和净化
提取:准确称取5.0g样品粉末于50mL塑料离心管中,加入1g NaCl和20mL 80%甲醇,涡旋5s,使充分分散于溶剂中,超声30min,再以300r/min的速度振荡30min。结束后,以5000r/min速度离心10min,准确移取2.0mL上清液加入磷酸盐缓冲液(PBS)稀释至25mL,充分摇匀,用玻璃微纤维滤纸过滤,收集滤液。
IAC净化:取上述滤液10mL加到预先平衡至室温的IAC柱,调节流速,使不超过1.0mL/min。待流干后,先用10mL PBS(pH7.2)淋洗,后用10mL超纯水淋洗。淋洗结束后,用吸耳球把柱内残余溶液尽可能挤掉,再用1.5mL甲醇分两次充分洗脱,用2.0mL塑料离心管收集洗脱液。洗脱液充分涡旋混匀后12000r/min离心3min,轻轻倒出上层液体于进样瓶中,供HPLC分析,外标法定量。进行空白实验时,取5mL纯水代替粉末样品,其余步骤相同。
1.4.2 样品溶液的测定
HPLC分析条件同1.3.2节。如所测溶液浓度超过标准曲线的线性范围,应将样品溶液稀释后再行测定。如超过IAC柱的最大亲和容量,则应降低取样量或者扩大稀释倍数后,重新用IAC制备样品溶液,再进行HPLC测定。
1.5 结果的计算
外标法定量,以保留时间定性,峰面积定量,则样品中OTA的浓度X/(μg/kg)为:
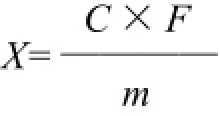
式中:C为待测溶液中OTA的质量浓度/(ng/mL);F为稀释倍数(F=25);m为称样质量/g。
2 结果与分析
2.1 液相色谱和样品净化条件的优化
2.1.1 OTA测定条件
[1]的方法,采用的流动相为甲醇-2%冰乙酸(65∶35,V/V),激发波长(λem)为333nm,发射波长(λem)=447nm,以Shiseido C18柱反相色谱柱作为分离柱,所得OTA色谱峰形对称,基线平稳(图1);经IAC柱净化的样品在此条件下,也得到较为理想的色谱峰。对HPLC仪器重复性进行了考察,对质量浓度10ng/mL的OTA溶液连续重复进样6次,所得峰面积的相对标准偏差(RSD)为0.748%,说明仪器性能稳定,自动进样的精密度高。

图1 标准溶液(A)和太子参样品(B)中OTA的色谱图Fig.1 Chromatograms of OTA in standard solution and in prince ginseng root extract
2.1.2 OTA的稳定性(再现性)实验
取同一OTA标准液系列,连续进样6d。OTA的日间RSD在1.70%~5.16%之间(表1),这说明仪器的再现性较好,也说明OTA化学性质较为稳定,可放置至少6d。

表1 OTA溶液的稳定性(再现性)实验Table 1 Stability evaluation of OTA solution during 6-day storage
2.1.3 样品净化条件的优化
2.1.3.1 亲和速度(流速)的优化
IAC柱装填有能与OTA特异性结合的抗体材料,这种特异性免疫亲和作用通常需要保持一定的时间,因此亲和过程中待测溶液流经IAC柱的速度可能影响到抗体对目标物OTA的亲和作用。实验比较了流速对两个品牌IAC柱的影响。由于LC tech公司的IAC柱内径较Vicam公司IAC柱大,容量多达3mL,因此通过固相萃取装置的调节阀来控制流速;Vicam公司IAC柱内径较小,流速较慢,因此采用加压的方式来提高流速。发现流速过快(大于1mL/min)时,两种柱的回收率为67.0%~70.9%,明显低于较低流速(小于1mL/min)时的回收率(88.4%~95.0%)。因此在上样时要控制流速,如果工作效率允许,尽量用较低的流速。
3.1.3.2 淋洗液对回收率的影响
不同的淋洗液对杂质的淋洗效果可能会有差异,从而影响IAC柱对OTA的特异性免疫亲和,同时也可能由于淋洗不充分而导致检测时有杂质干扰。比较纯水(10+10)mL、PBS缓冲液(10+10)mL、PBS 10mL+纯水10mL三种淋洗液对麦冬和龙眼肉中OTA测定的影响。结果发现,淋洗液对OTA的回收率无明显影响,回收率在85.5%~95.0%之间(表2)。考虑到基质的复杂多样,为保证有较好的淋洗效果,保护抗体的稳定性,又减少缓冲液中盐类进入色谱柱,通常缓冲液对抗体的保护能力更强,实验选择了PBS 10mL+纯水10mL这种3种淋洗方式。
2.2 方法学指标的考察
2.2.1 最低检出限
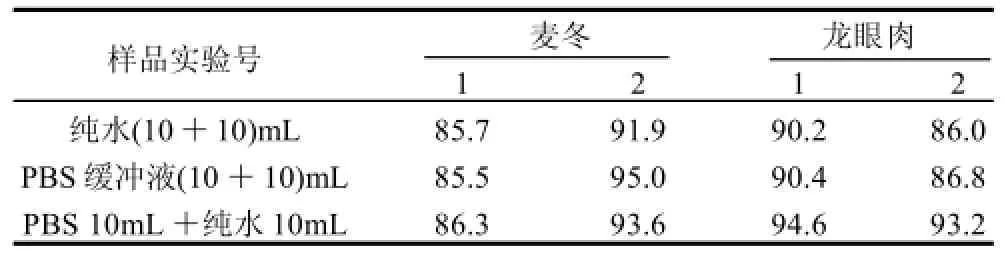
表2 淋洗液对样品OTA回收率的影响Table 2 Effect of selected eluents on recovery rate of OTA
将0.5ng/mL的标准溶液(OTA)稀释后再进样,得到信噪比为3时的OTA溶液浓度为0.20ng/mL。以5g样品计算,按照前述的稀释步骤,计算最低检出限为1.0μg/kg,满足了样品中OTA检测和污染控制的要求。
2.2.2 线性范围
在0.5~100ng/mL范围内,峰面积Y与OTA浓度X/ (ng/mL)呈线性关系,得到回归方程Y=9946.5X-8664.2,相关系数r=0.9995。在实际样品检测时,考虑到OTA标准液较为昂贵,同时减少操作中OTA的职业暴露风险,采用了0.5~10ng/mL的标准系列,折算为实际样品含量,相当于2.5~50μg/kg,完全满足日常检测的要求。
2.2.3 准确度
选择不含OTA的6种淀粉糖类药食两种食品样品(太子参、莲子、薏苡、泽泻、麦冬和龙眼),以此作为加标基质,加入不同体积的OTA(C=100ng/mL)标准溶液,使加标水平在1~8μg/kg之间,放置30min后,进行不同水平的加标回收率实验,每个水平重复实验3次,按照1.4节的样品制备和测定方法进行操作,结果见表3。在此水平下,除了泽泻4.0μg/kg加标水平回收率略高(116%)外,其余各类基质中OTA的平均回收率均在81.8%~107%之间,较为理想,满足欧盟对食品中OTA检测的质量控制要求[19](1~10μg/kg时,回收率在70%~110%),说明所用的两种商品IAC柱性能稳定、效果较好,操作过程容易控制。
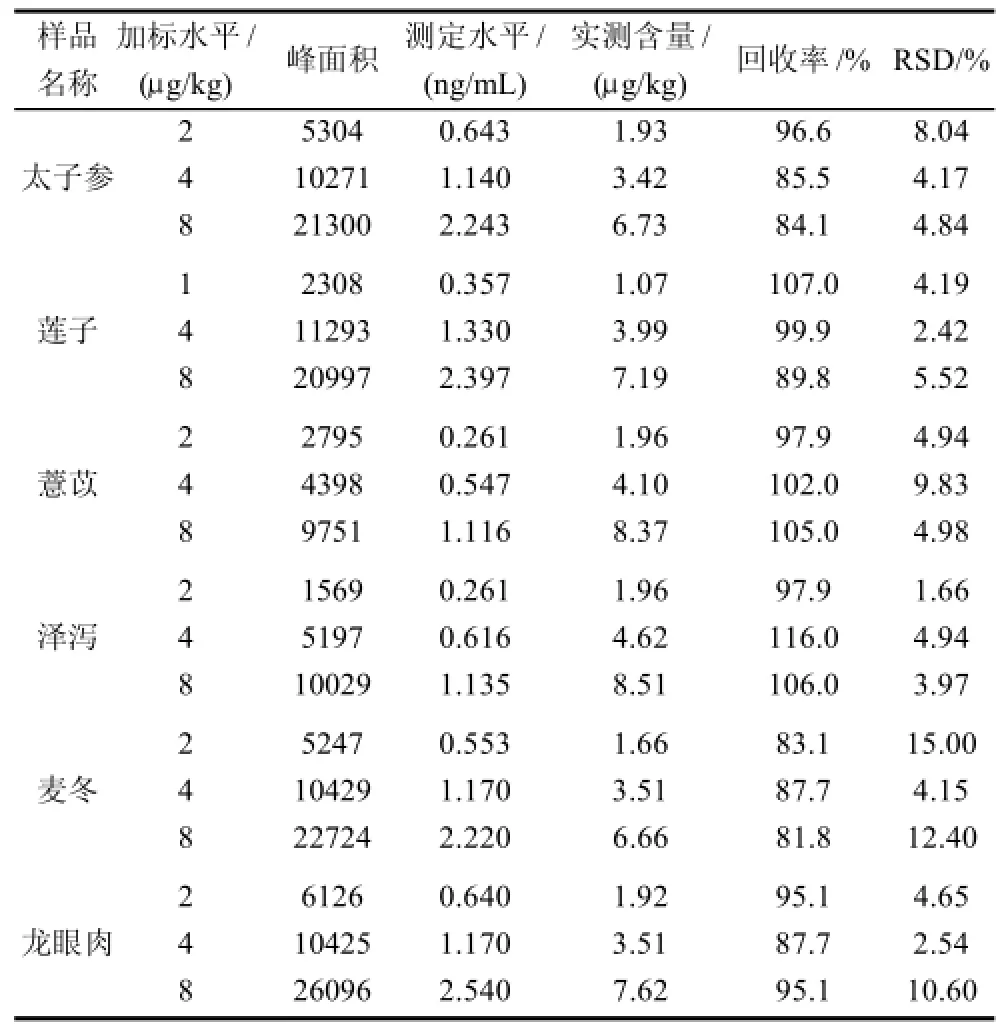
表3 某些基质中OTA的加标回收实验结果(n=3)Table 3 Mean recovery rates and RSDs for OTA in spiked food matrices (n=3)
2.2.4 精密度
为进一步考察样品制备直至HPLC分析整个过程中,OTA结果的一致性,对6种药食两用食品进行了重复加标实验(n=3),重复实验的RSD在1.66%~15.0%之间,精密度较为理想,优于欧盟对食品OTA检测的室内重复性RSD控制要求[19](1~10μg/kg时,RSDr<20%)。也说明商品OTA柱性能稳定,操作过程容易控制。
3 结 论
3.1 建立了6种淀粉糖类药食两用食品中赭曲霉毒素A的免疫亲和净化-液相色谱荧光检测方法,最低检出限为1.0μg/kg,平均回收率在81.8%~107%之间,RSD为1.66%~15.0%,各项性能指标满足该类食品中OTA检测和污染控制的要求。
3.2 免疫亲和柱净化效果理想,操作简便快速,易于掌握,重复性和再现性好,值得进一步推广用于其他品种食品和中赭曲霉毒素A的检测。
参考文献:
[1]李风琴. 赭曲霉毒素A分析方法进展[J]. 中国食品卫生杂志, 2004, 16(6)∶ 545-550.
[2]Codex Alimentarius Commission. Codex Stan 193—1995 Codex general standard for contaminants and toxins in foods[S].
[3]Commission of the European Communities. Commission Regulation (EC) No 1881/2006 Setting maximum levels for certain contaminants in foodstuffs[S]. 2006-12-06.
[4]GB 2715—2005 粮食卫生标准[S].
[5]Commission of the European Communities. Commission Regulation (EC) No 105/2010 Amending Regulation (EC) No 1881/2006 setting maximum levels for certain contaminants in foodstuffs as regards ochratoxin A[S]. 2010-02-05.
[6]KABELITZ L, SIEVERS H. Contaminants of medicinal and food herbs with a view to EU regulations[J]. Innovations in Food Technology, 2004(1)∶ 25-27.
[7]TRUCKSESS M, WEAVER C, OLES C, et al. Use of multitoxin immunoaffinity columns for determination of aflatoxins and ochratoxin A in ginseng and ginger[J]. Journal of AOAC international, 2007, 90(4)∶104-109.
[8]THIRUMALA-DEVI K, MAYO M A, REDDY G, et al. Occurrence of ochratoxin A in black pepper, coriander, ginger and turmeric in India[J]. Food Additives and Contaminants, 2001, 18(9)∶ 830-835.
[9]ARINO A, HERRERA M, ESTOPANAN G, et al. High levels of ochratoxin A in licorice and derived products[J]. International Journal of Food Microbiology, 2007, 114(3)∶ 366-369.
[10]PIETRI A, RASTELLI S, BERTUZZI T. Ochratoxin A and aflatoxins in liquorice products[J]. Toxins, 2010, 2(4)∶ 758-770.
[11]HERRERA M, HERRERA A, ARINO A. Estimation of dietary intake of ochratoxin A from liquorice confectionery[J]. Food and Chemical Toxicology, 2009, 47(8)∶ 2002-2006.
[12]SANTOS E A, VARGAS E A. Immunoaffinity column clean-up and thin layer chromatography for determination of ochratoxin A in green coffee[J]. Food Additives and Contaminants, 2002, 19(5)∶ 447-458.
[13]THIRUMALA-DEVI K, MAYO M A, REDDY G, et al. Production of polyclonal antibodies against ochratoxin A and its detection in chillies by ELISA[J]. Journal of Agricultural and Food Chemistry, 2000, 48 (10)∶ 5079-5082.
[14]褚庆华, 郭德华, 王敏, 等. 香辛料和藤蔓水果干中赭曲霉毒素A的测定[J]. 化学试剂, 2006, 28(10)∶ 597-600.
[15]TRUCKSESS M W, SCOTT P M. Mycotoxins in botanicals and dried fruits∶ a review[J]. Food Additives and Contamination, 2008, 25(2)∶181-192.
[16]TRUCKSESS M, WEAVER C, OLES C, et al. Determination of aflatoxins B1, B2, G1, and G2and ochratoxin A in ginseng and ginger by multitoxin immunoaffinity column cleanup and liquid chromatographic quantitation∶ collaborative study[J]. Journal of AOAC International, 2008, 91(3)∶ 511-523.
[17]MATISSEK R, RATERS M, HAREN W V, et al. Determination of ochratoxin A in liquorice products using HPLC based analytical methods. Part I∶ proficiency test of methods commonly used by the confectionary industry[J]. Mycotoxin Research, 2010, 26(2)∶ 93-99.
[18]GORYACHEVA I Y, DE SAEGER S, EREMIN S A, et al. Rapid all-inone three-step immunoassay for non-instrumental detection of ochratoxin A in high-coloured herbs and spices[J]. Talanta, 2007, 72(3)∶ 1230-1234.
[19]Commission of the European Communities. Commission Regulation (EC) No 401/2006. Laying down the methods of sampling and analysis for the official control of the levels of mycotoxins in foodstuffs[S]. 2006-02-23.
Determination of Ochratoxin A in Edible and Medicinal Foods by HPLC-Fluorescence Technology
FU Wu-sheng1,2,QIU Wen-qian1,ZHENG Kui-cheng1,LU Hua-dong1,GUO Bin3,YAN Xiao-bo4
(1. Fujian Center for Disease Prevention and Control, Fuzhou 350001, China;2. Teaching Base of Fujian Center for Disease Prevention and Control for Fujian Medical University, Fuzhou 350001, China;3. Fujian University of Traditional Chinese Medicine, Fuzhou 350001, China;4. College of Chemistry and Chemical Engineering, Fuzhou University, Fuzhou 350001, China)
Objective∶ To develop a method to determine ochratoxin A (OTA) in edible and medicinal foods using immunoaffinity column purification and high performance liquid chromatography (HPLC). Methods∶ Powdered samples were extracted with methanol-water (8∶2, V/V) sequentially by vortex mixing, ultrasonic treatment and shaking. The resulting extract was diluted with phosphate buffer solution, filtrated and cleaned up on immunoaffinity column (IAC) containing antibodies specific to OTA . The pooled eluate was separated on a C18 column (4.6 mm × 150 mm, 3μm) and detected by fluorescence detector (FLD). Results∶The limit of detection (LOD, RSN = 3) was 1.0μg/kg for OTA. An excellent linear relationship between peak area and OTA concentration was observed in the OTA concentration range of 0.5-100 ng/mL with correlation coefficient of 0.9995. The average recovery rates for OTA in prince ginseng root, lotus seed, coix seed, Radiχ Ophiopgonis, dried longan pulp and oriental water plantain rhizome spiked with OTA standard at a level of 1-8μg/kg were varied from 81.8%-107% with a relative standard deviation (RSD) of 1.66%-15.0% (n = 3). Conclusion∶ This method has the advantages of satisfactory results and high accuracy and precision and can meet the requirements of the Europe Union for the determination of OTA in food and feed, thus providing a suitable method for determining OTA edible and medicinal foods rich in starch or sugar.
foods;ochratoxin A;immunoaffinity cleanup;high performance liquid chromatography
O656.31
A
1002-6630(2011)14-0298-05
2010-08-19
福建省科技计划重点项目(2008Y0029)
傅武胜(1971—),男,副主任技师,博士,研究方向为污染物化学与风险评估。E-mail:fwsfqm@126.com
猜你喜欢
环境保护与循环经济(2021年7期)2021-11-02
考试与评价·高二版(2021年3期)2021-09-10
食品安全导刊(2021年20期)2021-08-30
基层中医药(2021年5期)2021-07-31
基层中医药(2021年12期)2021-06-05
基层中医药(2020年10期)2020-11-27
基层中医药(2020年9期)2020-11-27
数学物理学报(2020年5期)2020-11-26
天然产物研究与开发(2018年8期)2018-09-10
公民与法治(2016年14期)2016-05-17
