
一株新分离的肠杆菌噬菌体的快速遗传鉴定及其宿主识别基因的快速克隆
2011-02-10 01:20:24姜焕焕王盛李存刘大斌于长明安小平米志强陈建魁童贻刚
生物工程学报 2011年6期
姜焕焕,王盛,李存,刘大斌,于长明,安小平,米志强,陈建魁,童贻刚
1 军事医学科学院微生物流行病研究所 病原微生物生物安全国家重点实验室,北京 100071
2 军事医学科学院解放军307医院 临床检验科,北京 100071
Introduction
Bacteriophages are the most abundant entities in every ecosystem, and they can be found in all reservoirs populated by bacterial hosts, such as soil or the intestines of animals[1-3]. Since bacteriophages were discovered, they immediately attracted attention widely in the field of eradication of pathogenic bacterial infections. However, due to the limited knowledge regarding the biology of phages and how to use phage cocktails rationally, phages used in clinic were substituted by antibiotics. As a result of the frequent emergence of multi-drug resistant strains of many bacteria since the 1 950 s, phage therapy is receiving renewed interests as one of the viable alternatives to antibiotics[4]. Highly stringent host specificity is the major obstacle for phage therapy. The host specificity is mainly defined by the interaction of phage components and certain host surface structures. Many research groups engineered broader or changed spectrum phages based on knowledge of phage host recognizing proteins[5-7].
Escherichia coli, better known as E. coli was widely used in the research labs for a variety of different experiments, but some strains were pathogenic ones that cause disease even death. For example, E. coli was the cause of one third of childhood diarrhea cases in the developing countries[8], the recent emergence of E. coli with resistance to all antibiotics except to tigecycline and colistin conferred by New Delhi metallo-beta-lactamase 1 (NDM-1) potentially threaten public health greatly[9].
In order to develop Enterobacteria phage for prevention and treatment of infections especially by multi-drug resistant E. coli, we isolated an Enterobacteria phage IME08 from sewage, and then used a rapid and simple random PCR method to clone and sequence random fragments of its genome. BLAST results of these random fragments in NCBI website revealed it was a novel T4-like bacteriophage. The genus T4-like virus with phage T4 as the type specie is the most abundant genus in the field. Complete genomes of some individuals in this genus have been deposited into GenBank. T4-like bacteriophages have a genome of 150−200 kilobases. Gene 37 (g37) and gene 38 (g38) products (Gp37and Gp38) of T4-like bacteriophages are present at the tip of the long tail fibers and are supposed to be the determinants of host recognition[10-12].
To facilitate modifying the bacteriophage to finally broaden its host spectrum, we tried to determinate its host recognizing gene sequences. DNA sequence homology analysis showed that there was only a short conserved region of about 30 bp located at the 5' of g37, so we designed a primer in this region to directly sequence the ~180 kb phage genome, and fortunately we determined a short sequence of g37, the sequence obtained was subsequently used to design a nested primer. But when we used it to perform genome walking directly on the large phage genome, we could no long get any clear-cut sequence data of more than 300 bp. For this reason, we designed a novel PCR-based "genome jumping" strategy to clone and sequence bacteriophage host recognizing genes. This strategy allows unknown target DNA fragment of longer than 10 kb to be easily cloned without library construction, restriction digestion or adaptor ligation.
1 Materials and methods
1.1 Bacteria and growth conditions
The indicator strain used in this study was E. coli 8099 which was commonly used for disinfection agent evaluation[13]. Other E. coli K12 strains including DH5α, TOP10, JM109 and BL21, as well as clinical E. coli stains isolated from No. 307 Hospital (Beijing) were used to test lytic spectrum of the bacteriophage. All E. coli strains were kept at −80 °C in Luria-Bertani broth (LB) supplemented with 15% (V/V) glycerol, and these strains were routinely grown in LB broth or agar at 37 °C.
1.2 Bacteriophage isolation, purification and host range analysis
Bacteriophages were isolated from sewage using enrichment cultures. A total of 6 sewage samples from different hospitals were analyzed. Approximately 100 mL of filtrate (Millipore membranes, pore diameter 0.45 µm) was mixed with 50 mL of 3×LB and with 2 mL of overnight culture of E. coli strain 8099. The enrichment culture was incubated for at least 14 hours at 37 °C to allow amplification of bacteriophages, and then was centrifuged (10 min, 10 000×g, 4 °C). Bacteriophages in the supernatant were detected by the double-layered agar plate method. Briefly, 200 µL of the supernatant was mixed with 200 µL of overnight culture of E. coli strain 8099 and incubated at 37 °C for several minutes. 5 mL of melted top agar (soft LB with 0.7% agar) was added, and the mixture was poured onto a LB plate which was prewarmed. After incubating the plates at 37 °C overnight, the presence of bacteriophages in the form of plaques was examined.
To purify bacteriophage, plaques were picked and replaqued three times. Bacteriophage titration was performed by double-layered agar plate method described ahead. The titer of phages was calculated from the number of observed plaques and the known dilution factors. The host range of phage was initially screened by spotting 10 µL of SM buffer containing phages onto a lawn of an appropriate strain, and was further confirmed by plaque assay.
1.3 Bacteriophage genome extraction and rapid classification
Bacteriophage genetic material was extracted with the method described by Sambrook, et al[14], and then was digested with DNase I, RNase A and several restriction endonucleases respectively. After digestion, the fragments were analyzes by agarose gel electrophoresis. Furthermore, the genetic material was amplified by a two-step PCR using random primer and adaptor primer respectively[15], and DNA products approximately 1 kb in size were ligated with vector. Several appropriate clones confirmed by digestion with restriction endonucleases were sequenced, and DNA database searches were done by Blast on NCBI website to analyze its probable taxonomic status[16].
1.4 Electron microscopy of bacteriophages
The morphology of bacteriophage IME08 was examined by Electron microscopy, following the protocol developed by Williams[17]. Briefly, 50 µL of purified viral stock solution was fixed by addition of 50 µL of 0.5% glutaraldehyde in 4% paraformaldehyde. The bacteriophage solution was the placed on a carbon coated copper grid. After 30 min settlement, excess liquid was removed and the grid was allowed to dry. A drop of 2% phosphotungstic acid was then added and after 2 min the excess was removed with a piece of filter paper. After the grid was dried, it is examined by TEM (Philips, TECNAI-10) at an acceleration voltage of 80 kV.
1.5 Determination of host-recognizing genes sequence
1.5.1 The primary PCR
The primary PCR reaction was performed in a 50 µL solution containing 100 ng of genomic DNA, 250 µmol of each dNTPs, 0.4 µmol of primer-1 and primer-3 (Table 1), and 2.5 units of TransStart Fast Pfu DNA Polymerase (TansGen). The process included the following steps performed on a GeneAmp PCR system 2400 (Perkin Elmer): 2 min denaturation at 95 °C, five cycles of regular PCR amplification (10 s at 95 °C, 20 s at 45 °C, 2.5 min at 72 °C), two cycles of low annealing-temperature PCR (10 s at 95 °C, 20 s at 30 °C, 2.5 min at 72 °C), and 30 cycles of regular PCR amplification again (10 s at 95 °C, 20 s at 45 °C, 2.5 min at 72 °C). The reaction was ended with a final elongation step of 7 min at 72 °C.

Table 1 Primers used in the primary and secondary PCR
1.5.2 The secondary PCR
Ten microliters of purified DNA products of the primary PCR (>5 kb) were used as template in the secondary PCR. Components of the secondary PCR were the same as these of the primary PCR except that primer-1 and primer-3 were replaced by primer-2 and primer-4 (Table 1). Following one denaturation step of 2 min at 95 °C, the reaction proceeded with 35 cycles of regular PCR amplification (10 s at 95 °C, 20 s at 60 °C, 2.5 min at 72 °C) and was ended with a final elongation step of 7 min at 72 °C.
1.5.3 Sequence determination and characterization
The PCR products were cloned and a clone with insertion of about 5 kb was sequenced from both directions using Sanger method. The determined ORFs and the deduced amino acids of g37 and g38 were analyzed with Blastn and Blastp on NCBI website respectively to search for homologous entries. The sequences with high identities were downloaded and aligned with each other locally using DNAStar to detect mutual sequence similarities.
2 Results and discussions
2.1 Isolation and characterization of IME08
Phage IME08 was isolated using E. coli strain 8099 as a host. Spot tests showed that the phage IME08 could efficiently infect other K12 strains (including DH5α, TOP10, JM109 and BL21). We tested IME08 on 13 clinical E. coli strains isolated from Beijing No. 307 Hospital, and 6 of these strains produced plaque, suggesting that these strains were permissive hosts for bacteriophage IME08. The genetic material digested with DNase I, RNase A and restriction endonucleases respectively indicated that it was double stranded DNA. Four clones carrying random DNA fragments were sequenced, and Blast results showed that phage IME08 random fragments were in close relation with coliphage T4 (94% identity), JS98 (95% identity), JS10 (95% identity) and RB14 (94% identity) respectively. Since all of above strains belonged to T4-like virus, we supposed phage IME08 was a novel T4-like phage. Negative stain electron micrograph showed the phage particles had an icosahedral capsid and a contractile tail with fibers. The capsid was of approximately 60 nm in width and 80 nm in length, and the length of the tail was around 55 nm, which was typical for T4-like bacteriophage.
2.2 Principle of the genome jumping strategy
The principle of this strategy is illustrated in Fig. 1. The primary PCR started with the high-stringency amplification step which allows the gene-specific primer to drive the linear amplification of singlestranded specific DNA of various sizes. The length is determined by the capacity of the thermostable DNA polymerase and the extension time. And then the low-temperature annealing step lets the random primer to bind to nonspecific sites to synthesize singlestranded random DNA molecules. This is followed by another high-stringency amplification step which leads to the amplification of both specific and nonspecific sequences. Since short DNA molecules are more efficiently amplified than the longer ones, gel separation of the primary PCR products will significantly reduce the amount of the undesired short DNA molecules and avoid the preferential amplification of short fragments in the following secondary PCR. The nested gene-specific primer and the adaptor primer further increase the ratio of the specific DNA molecules and the second gel separation makes the final products contain mainly specific DNA of desired length. For screening of the transformants, the restriction site included in the adapter primer offers an advantage for distinguishing specific and nonspecific clones.
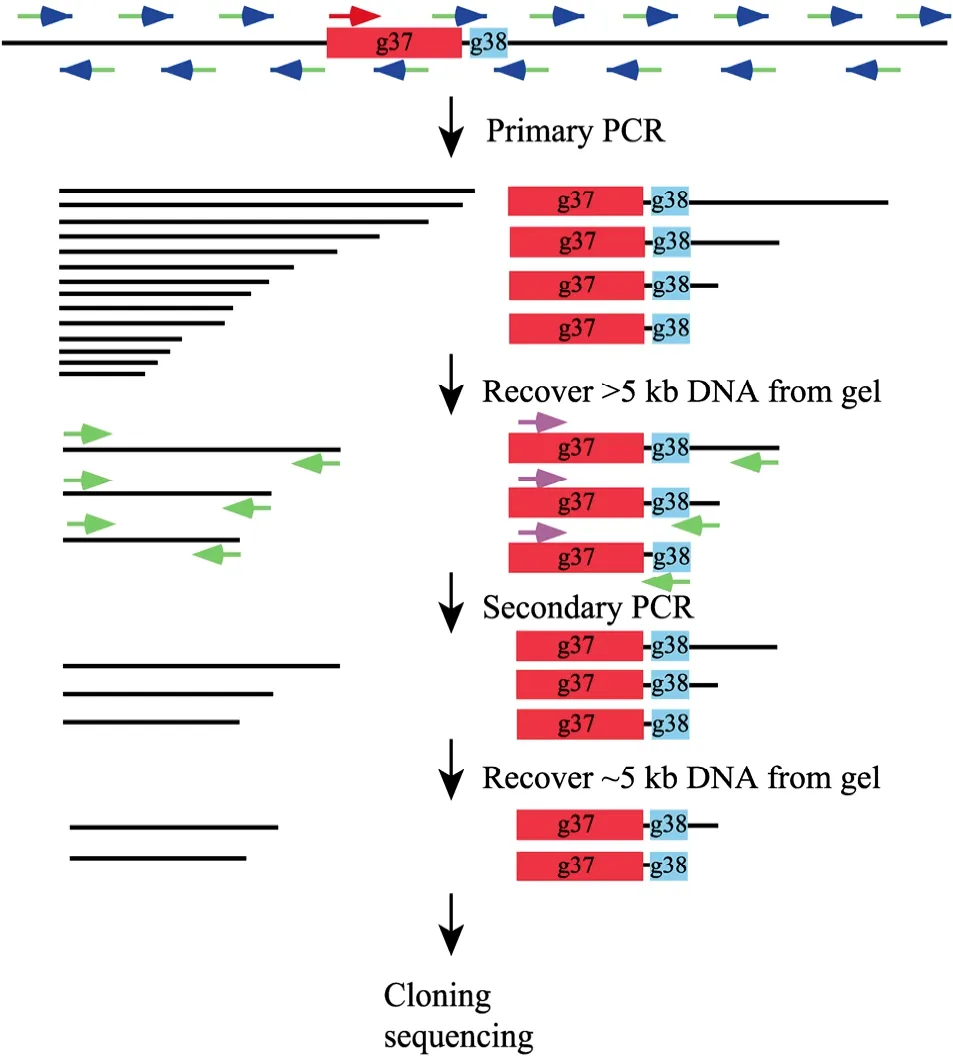
Fig. 1 The schematic diagram of the "genome jumping" strategy. Refer to the text in section "Principle of the genome jumping strategy" for the detailed explanation of the procedures. Briefly, the primary PCR with a gene-specific primer and a random primer generates both specific and nonspecific sequences of various sizes. The first DNA extraction step excluded small molecules to reduce the invalid templates. The secondary PCR with a specific nested primer and the adaptor-primer produces mainly specific products and the second gel purification step ensure the recovery of desired fragments of specific size. The thin lines represent the bacteriophage genomic sequences. The red boxes and green boxes represent g37 and g38, respectively. PCR primers are shown by arrows, with the red and pink arrows represent gene-specific outside and nested primers respectively. The blue arrows and its green tails indicate the random hexamers and the attached adaptor-sequence. The green arrows represent the adaptor-primer.
2.3 Amplification and cloning of phage IME08 g37 and g38
The primary PCR products were separated on 1% (W/V) agarose gel electrophoresis. As show in Fig. 2A, DNA molecules of various sizes were generated in the primary PCR but most of them were smaller than 5 kb in size. In order to enrich the valid DNA molecule templates and improve the production of specific molecules of desired length, the DNA molecules that range from 5 kb to 10 kb in size were excised from the gel and purified with EasyPure Quick Gel Extraction Kit (TransGen). As expected, in the secondary PCR amplification, the amount of longer DNA molecules was much more than that of the primary PCR even though a few distinct DNA bands smaller than 3 kb were exhibited (Fig. 2B). These small DNA products (<5 kb) may be resulted from adaptor-primer amplifying nonspecific templates and the preferential amplification of short fragments. After gel fractionation, the DNA molecules of about 5 kb in size were extracted from the gel and ligated with a blunt cloning vector pEASYBlunt Simple (TransGen). This cloning vector is 4 kb in size and does not contain any common restriction site like Sac I. The ligation mixture was then transformed into Escherichia coli strain Trans1-T1 Phage Resistant Chemically Competent Cell (TransGen) and the transformants were grown on LB plates supplemented with ampicillin (100 µg/mL). The clones were analyzed by Sac I restriction digestion and the results demonstrated that most of the transformants were gene-specific clones (only one Sac I site) (Fig. 3). Some of the insertions were much smaller than 5 kb, probably due to poor gel separation and carry-over contamination of smaller DNA fragments, as well as better stability and higher transformation efficiency of smaller plasmids.
2.4 Characteristics of IME08 g37 and g38
For g37 of IME08, the nucleotide sequence showed 61% to 69% identity compared to T4, K3 and T2 while the deduced amino acids (1257 amino acids) showed 43% to 59% identity (Table 2). About 50 aminoterminal residues of Gp37 are almost completely conserved among T4-like virus genus. Most of the variation among Gp37 of these T4-like bacteriophages occurs near the carboxyl-terminal end. Like Gp37 of T2 and K3, Gp37 of IME08 does not contain the tripeptide repeats His-Ser-His or His-Thr-His which were believed to be the determinants of host recognition[10].

Fig. 2 Analysis of the primary and secondary PCR products. The PCR products were separated on 1% (W/V) agarose gel electrophoresis and visualized with ethidium bromide. (A) Primary PCR. (B) Secondary PCR. S: PCR products; M: 1 kb DNA ladder.

Fig. 3 Transformants analysis with Sac I Digestion. The Sac I digestion products of randomly selected clones are analyzed by 1% (W/V) agarose gel electrophoresis stained with ethidium bromide (lanes 1-11). The cloning vector pEasy-Blunt Simple (TransGen) is 4 kb in size and contains no Sac I site. Since the adaptor-primer contains a Sac I site, the clones of random insertion generated by two adaptor-primers exhibit at least two bands (a vector band and an insertion band, lane 7 and 8), while the gene-specific clones (lane 1-6, 9-11) display only one band. The sizes of insertions range from 1 kb to 5 kb. M: 1 kb DNA ladder.

Table 2 Sequence identities between g37 of IME08, T2, T4 and K3 respectively
g38 of IME08 showed 54% to 80% nucleotide sequence identity with its counterpart from T4, K3 and T2. The deduced acid amino sequence of Gp38 (262 amino acids) showed the highest identities with the corresponding products of T2 (88%) (Table 3). Gp38 shows high frequency of alanine (10.62%) and extremely high frequency of glycine (22.34%), which is coincident with Gp38 from T2 and K3[11].

Table 3 Sequence identities between g38 of IME08, T2, T4 and K3 respectively
Based on the comparisons of nucleotide and amino acid sequences, we conclude that phage T2 is the nearest match of this novel bacteriophage IME08. We tried to build a phylogenetic tree for IME08 g37 and g38, but there are only a few entries in GenBank which display overall sequence similarities and most of them (except T2, K3, and T4) share little sequence identities with IME08. The current data of IME08 g37 and g38 enriched the information about the host-recognizing gene of bacteriophage and can be utilized to modify the host range of the most abundant lytic T4-like phages for potential clinical application. The g37 and g38 sequences described in present study have been deposited into GenBank with accession numbers GU208851 and GU208852 respectively.
3 Conclusion
In the past several decades, antibiotics were used as the main treatment of bacterial infections, but with the surprisingly increasing emergence of multi-drug resistant bacteria, the effectiveness of antibiotics is under suspicion. As the natural bacteria killers, bacteriophages display more advantages than antibiotics to the treatment of bacterial infections especially of multi-drug resistant bacteria. Many studies had focused on application of bacteriophages as an alternative to antibiotics in animal or clinical trials[18-20]. But compared with the numerous bacteriophages existing in environment, the isolated phages are extremely limited, and only a few of them have been studied in detail. Here we isolated a novel Enterobacteria phage IME08 from hospital sewage, and confirmed it was double-stranded DNA phage by digesting its genetic material with DNase I, RNase A and several restriction endonucleases respectively. BLAST results of random fragments generated by a random PCR cloning method revealed that it belonged to T4-like virus. We subsequently determined the host recognizing genes (g37 and g38) sequence with a PCR-based "genome jumping" protocol based on highly conserved region at 5' terminus of g37 from four other T4-like Bacteriophages (T4, JS98, T2 and K3). This molecular biological method enabled us to efficiently determine the sequence of the genes of interest based on very limited conserved sequence information. Compared with other existing PCR-base genome walking methods, this protocol does not require genome breakup, DNA restriction digestion, library construction or adaptor ligation. It also does not require special reagents or commercial kits except for the ordinary reagents such as thermostable DNA polymerase and DNA primers, which makes this strategy readily applicable for cloning purposes in individual laboratories. Another important advantage of this protocol is that it can clone a target sequence of desired length. So if necessary, one can precisely pinpoint a target sequence within a range of several kilobases. The largest length of the DNA fragments to be cloned will depend on the amplification ability of the thermostable DNA polymerase and the capacity of the cloning vector, which at present could be 15 kb to 20 kb. Thus, with this protocol, we can easily "jump" 10−20 kb every time in an un-sequenced genome about which only limited or incomplete sequence information is available.
[1] Bettarel Y, Bouvy M, Dumont C, et al. Virus-bacterium interactions in water and sediment of West African inland aquatic systems. Appl Environ Microbiol, 2006, 72(8): 5274−5282.
[2] Kutter E, Sulakvelidze A. Bacteriophages Biology and Applications. Boca Raton: CRC Press, 2005.
[3] Wommack KE, Colwell RR. Virioplankton: viruses in aquatic ecosystems. Microbiol Mol Biol Rev, 2000, 64(1): 69−114.
[4] Wright A, Hawkins CH, Änggård EE, et al. A controlled clinical trial of a therapeutic bacteriophage preparation in chronic otitis due to antibiotic-resistant Pseudomonas aeruginosa; a preliminary report of efficacy. Clin Otolaryngol, 2009, 34(4): 349−357.
[5] Montag D, Schwarz H, Henning U. A component of the side tail fiber of Escherichia coli bacteriophage λ can functionally replace the receptor-recognizing part of a long tail fiber protein of the unrelated bacteriophage T4. J Bacteriol, 1989, 171(8): 4378−4384.
[6] Tétart F, Repoila F, Monod C, et al. Bacteriophage T4 host range is expanded by duplications of a small domain of the tail fiber adhesin. J Mol Biol, 1996, 258(5): 726−731.
[7] Yoichi M, Abe M, Miyanaga K, et al. Alteration of tail fiber protein gp38 enables T2 phage to infect Escherichia coli O157: H7. J Biotechnol, 2005, 115(1): 101−107.
[8] Albert MJ, Faruque SM, Faruque AS, et al. Controlled study of Escherichia coli diarrheal infections in Bangladeshi children. J Clin Microbiol, 1995, 33(4): 973−977.
[9] Kumarasamy KK, Toleman MA, Walsh TR, et al. Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: a molecular, biological, and epidemiological study. Lancet Infect Dis, 2010, 10(9): 597−602.
[10] Riede I, Drexler K, Eschbach ML, et al. DNA sequence of the tail fiber genes 37, encoding the receptor recognizing part of the fiber, of bacteriophages T2 and K3. J Mol Biol, 1986, 191(2): 255−266.
[11] Riede I, Drexler K, Eschbach ML, et al. DNA sequence of genes 38 encoding a receptor-recognizing protein of bacteriophages T2, K3 and of K3 host range mutants. J Mol Biol, 1987, 194(1): 31−39.
[12] Riede I, Eschbach ML, Henning U. DNA sequence heterogeneity in the genes of T-even type Escherichia coli phages encoding the receptor recognizing protein of the long tail fibers. Mol Gen Genet, 1984, 195(1/2): 144−152. [13] Yuan QX, Zhang WF, Zhong RB, et al. Comparative resistance of different standard test bacteria tovailable chlorine. Chin J Disinfect, 2001, 18(2): 90−92.
[14] Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 3rd ed. New York: Cold Spring Harbor Laboratory Press, 2001.
[15] Djikeng A, Halpin R, Kuzmickas R, et al. Viral genome sequencing by random priming methods. BMC Genomics, 2008, 9(1): 5.
[16] Wang S, Jiang HH, Chen JK, et al. Isolation and rapid genetic characterization of a novel T4-like bacteriophage. J Med Coll PLA, 2010, 25(6): 331−340.
[17] Williams BD, Carter CB. Transmission Electron Microscopy: A Textbook for Materials Science. New York: Springer, 2009.
[18] Fu WL, Forster T, Mayer O, et al. Bacteriophage cocktail for the prevention of biofilm formation by Pseudomonas aeruginosa on catheters in an in vitro model system. Antimicrob Agents Chemother, 2010, 54(1): 397−404.
[19] Kumari S, Harjai K, Chhibber S. Evidence to support the therapeutic potential of bacteriophage Kpn5 in burn wound infection caused by Klebsiella pneumoniae in BALB/c mice. J Microbiol Biotechnol, 2010, 20(5): 935−941.
[20] Kutter E, De Vos D, Gvasalia G, et al. Phage therapy in clinical practice: treatment of human infections. Curr Pharm Biotechnol, 2010, 11(1): 69−86.
猜你喜欢
中华养生保健(2020年3期)2020-11-16 00:53:20
生物技术通讯(2017年2期)2017-04-12 19:24:01
生物技术通讯(2017年1期)2017-04-10 20:32:50
分子诊断与治疗杂志(2017年2期)2017-03-30 02:46:58
生物学教学(2016年3期)2016-04-10 13:06:09
中国卫生产业(2016年18期)2016-02-06 11:21:57
中国卫生产业(2015年29期)2015-06-05 03:49:06
江苏卫生事业管理(2014年2期)2014-02-28 01:59:37
现代检验医学杂志(2014年3期)2014-02-02 02:42:45
中国比较医学杂志(2010年10期)2010-02-11 15:20:03
