
PCR依赖型方法构建高质量酵母基因突变文库
2011-02-09 09:31王睿喻晓蔚徐岩郅岩孔宇
生物工程学报 2011年9期
王睿,喻晓蔚,徐岩,郅岩,孔宇
1 江南大学 工业生物技术教育部重点实验室,无锡 214122
2 江南大学医药学院,无锡 214122
酶的体外定向进化属于蛋白质的理性设计,它无需事先了解酶的空间结构和催化机制,只需人为地创造特殊的条件,模拟自然进化机制 (随机突变、重组和自然选择),在体外改造酶基因,并定向选择出所需性质的突变酶[1]。酶的体外定向进化技术极大地拓展了蛋白质工程学的研究和应用范围,特别是能够解决理性设计所不能解决的问题,为酶的结构与功能研究开辟了崭新的途径,并且正在工业、农业、食品和医药等领域逐渐显示其生命力[2-6]。
酵母是表达外源基因的最重要的表达系统之一。酵母作为单细胞生物,在生产和操作上具有优越于细菌表达系统的易于大规模扩增的特点,又具有真核细胞对蛋白质的翻译后加工及修饰的作用[7-8]。酵母表达系统的表达载体主要分为2种,即整合型载体和附加型载体。整合型载体不含自主复制序列,转化入酵母后整合到酵母宿主菌的染色体上,具有稳定性好的优点,尤其适合于工业化生产。而附加型载体能够在酵母体内自主复制,达到较高的拷贝数,但是在非选择条件下多不稳定[9]。
定向进化的研究中常用大肠杆菌为宿主构建基因突变文库,而酵母使用较少。而对于在大肠杆菌表达体系中表达效果不好的酶,就需要尝试其他表达体系。前期研究表明华根霉脂肪酶在大肠杆菌中表达无法进行翻译后修饰,比活很低,而无法体现出脂肪酶应有的酶学性质,筛选困难[10];而该脂肪酶在毕赤酵母中具有较高的表达量,比活与野生型相当,能够反应出脂肪酶应有的酶学性质[11-12]。但是利用亚克隆法构建酵母突变文库过程繁琐,步骤包括:目的基因突变;突变基因与表达载体的酶切处理;重组质粒构建;重组质粒转化大肠杆菌进行大量扩增;重组质粒的提取及酶切线性化;转化酵母,获得基因突变文库。上述过程繁琐、低效、耗时耗力,极大地损失了突变基因的容量和丰度,而目的转化子需要从成千上万个转化子中才能筛选得到,因此上述方法不能满足构建酵母整合型基因突变文库的需要。
本研究建立的构建酵母突变文库的新方法与亚克隆法一样,都是以构建的重组表达质粒能够与毕赤酵母基因组发生同源重组为基础,获得外源基因在毕赤酵母中的表达。但是两者构建重组表达质粒的过程和原理不同。亚克隆法是在体外构建表达盒:需要在体外获得可以与酵母基因组进行同源重组的完整片段 (即将目的基因经过 PCR扩增,酶切,与酶切后的表达载体连接获得重组表达质粒,再扩增重组质粒,对重组质粒进行线性化),才可以将该片段转化进入酵母,与酵母基因组同源重组后获得突变文库。而新方法是在体内自主构建表达盒:不需要在体外获得完整的片段,只需要分别扩增出目的基因和表达载体,将其混合转化毕赤酵母,基因片段和载体片段上的同源臂可以在酵母体内同源重组形成与亚克隆法体外获得的完全相同的完整片段,再与酵母基因组同源重组,获得突变文库。
1 材料与方法
1.1 材料
1.1.1 酶和试剂
限制性内切酶,T4 DNA连接酶,Taq DNA聚合酶,PCR试剂 (TaKaRa公司),引物,DL2000 DNA Ladder Maker,E10000 DNA Ladder Maker,醋酸锂/山梨醇 (上海生工有限公司),YNB,G418,DTT (BIOSHARP),琼脂糖凝胶DNA回收试剂盒,PCR产物纯化试剂盒 (天根生化科技 (北京) 有限公司),Plasmid Mini Kit I (OMEGABIO-TEK),其余试剂均为国产或进口分析纯。
1.1.2 菌株和质粒
大肠杆菌Escherichia coli JM109、巴斯德毕赤酵母 Pichia pastoris GS115和华根霉;Rhizopus chinensis CCTCCM201021;由本实验室保存;载体pPIC9K购自Invitrogen公司;携带华根霉脂肪酶基因proRCL的重组质粒pPIC9K-proRCL由本实验室构建。
1.1.3 培养基
YPD:1%酵母膏,2%蛋白胨,2%葡萄糖。
MD:1.34%YNB,4×10−5%生物素,2%葡萄糖。
YPD-G418 (0.5~3 g/L):1%酵母膏,2%蛋白胨,2%葡萄糖,0.5~3 g/L G418。
BMMY:1%酵母膏,2%蛋白胨,0.5%甲醇,0.1 mmol/L磷酸缓冲液 (pH 6.0),1.34% YNB,4×10−5生物素。
BMMYA’:1%酵母膏,2%蛋白胨,2%甲醇,0.1 mmol/L磷酸缓冲液 (pH 6.0),1.34% YNB,4×10−5生物素,2%琼脂。
1.2 方法
1.2.1 proRCL的易错PCR扩增
为了使载体片段 pPIC9K与扩增基因片段在电转后插入酵母基因组时能够有效地同源重组,基因片段与PIC9K必须具有不小于40 bp的同源臂。以重组质粒 pPIC9K-proRCL中 proRCL上下游的PIC9K序列为模板,分别设计目的片段的上游引物66 bp F和下游引物60 bp R (表1)。其中,上、下游引物分别与pPIC9K有66 bp和60 bp的重叠。
50 μL易错PCR体系如下:5 μL 10×易错PCR缓冲液 (500 mmol/L KCl,70 mmol/L MgCl2,100 mmol/L Tris-HCl (pH 8.3),0.1% (W/V,明胶));0.5 mmol/L dATP/dGTP,2.5 mmol/L dCTP和dTTP;引物各40 pmol;MgCl27 mmol/L;MnCl20.3 mmol/L;Taq DNA聚合酶2.5 U。PCR扩增条件:94 ℃预变性3 min;94 ℃ 1 min ,59 ℃ 1 min,72 ℃ 2 min,30个循环;72 ℃延伸10 min。
易错PCR扩增产物命名为promRCL,经DNA纯化试剂盒纯化。
1.2.2 表达载体 pPIC9K的扩增
载体处理的一般方法是对载体进行酶切处理,再与目的基因连接。对 pPIC9K进行双酶切后,可得到一段大小为9 226 bp的片段[6]。可双酶切操作耗时、繁琐、低效,不能满足快速高效制备载体的要求。

表1 本研究中用到的引物及其序列Table 1 The primers utilized in this study and primer sequences
本研究实现了载体的PCR制备,无需酶切。由于需要扩增得到末尾不加“A”的载体片段,且需要保证扩增片段的高保真性,选择Prime STAR聚合酶(SP) 进行扩增。
以质粒 pPIC9K为模板,以引物 9K-shortF和9K-shortR (表1) 扩增得到9K-short (2 000 bp) 的短片段;同样以引物9K-longF和9K-longR扩增得到9K-long (7 226 bp) 的长片段。
其中,9K-long的PCR扩增程序使用两步法:98 ℃预变性1 min;98 ℃ 10 s,68 ℃ 7.5 min,30个循环;68 ℃延伸10 min,可达到较好的扩增效果。
将扩增片段9K-long和9K-short利用纯化试剂盒进行纯化后备用。
1.2.3 基因与载体同源臂长度对建库的影响
为考察不同长度同源臂对整个建库过程的影响,设计不同长度的引物 9K-longF/R 和9K-shortF/R,使目的基因与载体间同源臂的长度分别位于以下区间:25~40 bp,40~70 bp和>70 bp。将上述目的基因与载体分别电转,获得突变文库,每个文库随机挑选10个菌株来测定其阳性重组率。
1.2.4 突变基因proRCL扩增片段和表达载体pPIC9K混合
将9K-long和9K-short片段以等摩尔混合,再与突变基因promRCL片段按照摩尔比10∶1,3∶1,1∶1,1∶3和1∶10的梯度混合均匀,使各梯度基因总量相同。于真空干燥箱浓缩至体积<1 μL。
1.2.5 载体与基因混合物的电转化
对电转化过程中各参数进行优化[13]。电转化过程按照取混合均匀的片段加入至80 μL感受态细胞中,转至0.2 cm冰预冷的电激杯。冰上放置5 min,电压1 500 V,电容25 μF,电阻200 Ω,进行电击。立刻加入1 mL 1 mol/L山梨醇复苏,30 ℃培养1 h。将转化液涂布于MD平板上,30 ℃培养2 d,得到突变文库。
1.2.6 阳性转化子的确定
阳性转化子可以表达脂肪酶,所以可以在含Fast-blue RR顶层琼脂的平板上清晰显色,另外,经PCR验证,外源 DNA和载体片段应重组为正确的表达盒并插入到酵母基因组内。
将 MD平板上的单菌落用灭菌牙签挑至YPD-G418 (0.5 g/L),30 ℃培养2 d。不带有基因的9K载体与酵母基因组发生同源重组产生的重组菌也可以在 G418抗性平板上生长。对 MD平板和YPD-G418平板进行计数。
阳性率=G418抗性平板上的单菌落数/MD板上的单菌落数。
进行阳性重组率测定实验时,使用常规PCR而非易错PCR扩增产物,以消除基因突变带来的酶活的影响。将 MD平板上的单菌落用灭菌牙签挑至BMMYA’板上,诱导5 d后,Fast-blue RR顶层琼脂法染色,2 min内能够显出明显棕黑色的菌株为重组阳性菌 (脂肪酶可以水解萘酯,水解产物与染料结合显色)。重组阳性菌是指带有脂肪酶基因 proRCL的 9K载体片段与酵母基因组发生同源重组产生的菌株。
重组率=所有BMMYA’板上的单菌落中可以诱导表达产脂肪酶的单菌落数/YPD-G418板上的单菌落数。
若目的基因为其他基因,应选择其相应的酶活测定方法确定阳性重组率。
为了验证电转时外源 DNA与载体片段是否同源重组为正确的表达盒并插入到酵母基因组中,提取筛选目的菌株的基因组,以5′AOX和3′AOX为引物 (表1) 进行扩增,产物送往上海生工生物有限公司测序。
1.2.7 PDM构建的重组菌的酶活测定
pNPP法测定脂肪酶酶活[14]。比较PDM构建的突变文库与亚克隆方法构建的突变文库,考察库容量及筛选得到的突变株的酶活、表达量、热稳定性等其他酶学性质。
1.2.8 PDM构建的突变文库的筛选
根据筛选目的,选择合适的筛选方法对PDM构建的突变文库进行筛选。文中分别筛选得到了酶活提高,酶表达量提高以及酶热稳定性提高的突变株。
筛选酶活提高突变株的方法为:用灭菌牙签将 MD平板上生长出的 His+转化子复制到YPD和 BMMY平板的相同位置,同时将对照菌GS115/pPIC9K-proRCL Mut+接种至BMMY平板上。30 ℃培养2 d后,每12 h向BMMY平皿盖上补加200 µL甲醇诱导重组脂肪酶表达。诱导2~3 d。向酶活测定底物pNPP中加入0.6%琼脂,混合均匀后,倒于BMMY平板形成顶层琼脂。2 min内出现明显黄色的菌株为初筛目的菌株。相同条件下,对照菌GS115/pPIC9K-proRCL Mut+不能显示出明显黄色[14]。再利用 96孔板发酵法对初筛目的菌株进行进一步筛选。
筛选酶表达量提高突变株的方法为:用灭菌牙签将MD平板上生长出的His+转化子复制到 YPD-G418 (0.5 g/L)、YPD-G418 (1 g/L)、YPD-G418 (2 g/L)、YPD-G418 (3 g/L) 平板的相同位置,能在YPD-G418 (0.5 g/L) 平板上生长良好,在另 3块高浓度的 YPD-G418平板或其中一块上,也生长到和YPD-G418 (0.5 g/L) 平板上几乎一样的大小,即为表达量提高的初筛目的菌株。再利用 96孔板发酵法对初筛目的菌株进行进一步筛选。
筛选酶热稳定性提高突变株的方法为:用灭菌牙签将 MD平板上生长出的 His+转化子复制到BMMYA’平板上和YPD平板的相同位置,同时将对照菌 GS115/pPIC9K-proRCL Mut+接种至 BMMYA’平板上。30 ℃生长并诱导4~5 d后,平板经脂肪酶致死温度65 ℃处理1 h,冰浴、室温平衡后,向平板中倾入Fast-blue RR顶层琼脂。2 min内出现明显褐色的菌株为初筛目的菌株。相同条件下,对照菌不能显示出明显褐色。再利用96孔板发酵法对初筛目的菌株进行进一步筛选。
2 结果与分析
2.1 proRCL的易错PCR扩增及载体片段pPIC9K的扩增
引物66 bpF/60 bpR经error-prone PCR扩增得到大小为1 218 bp (1 092 bp+66 bp+60 bp) 的proRCL基因片段 (扩增片段上下游与载体含有 60 bp和66 bp的重叠序列) (图 1中 First PCR)。以质粒pPIC9K为模板,9K-longF/R和9K-shortF/R为引物分别扩增得到两段大小分别为7 226 bp和2 000 bp的片段 (图1中Second/Third PCR)。
2.2 载体线性化对转化效率的影响
亚克隆方法中,电转化重组表达质粒pPIC9K-proRCL前,需要对其进行酶切线性化,才能电转化并与酵母基因组进行同源重组。环状质粒无法重组成功[15]。PDM方法中,也考察了载体线性化对重组效率的影响。PCR扩增载体时,设计了 2组实验。
实验 1:以 pPIC9K-proRCL为模板,利用 9K-shortF和 9K-longF一组引物扩增出长为 9 226 bp (2 000 bp+7 226 bp) 的片段;实验2:利用9K-shortF/9K-shortR和9K-longF/9K-longR两组引物分别扩增长为2 000 bp和7 226 bp的片段。
实验1对应于亚克隆方法中不对表达质粒进行酶切线性化;实验2对应于亚克隆方法中对表达质粒进酶切线性化。保证2组实验的基因及载体的量相同,均为基因5 µg (0.5 nmol),载体片段 5 nmol,混匀并浓缩。此时混合物浓度约为10 g/L。将80 µL感受态加入上述2管DNA混合物中,电转化。
转化结果为:实验1:MD板上单克隆数为2个,均为假阳性;实验2:4 000~5 000个,阳性率达95%。此结论说明利用亚克隆或PDM的方法构建酵母突变文库时,表达载体的线性化都是必需的。
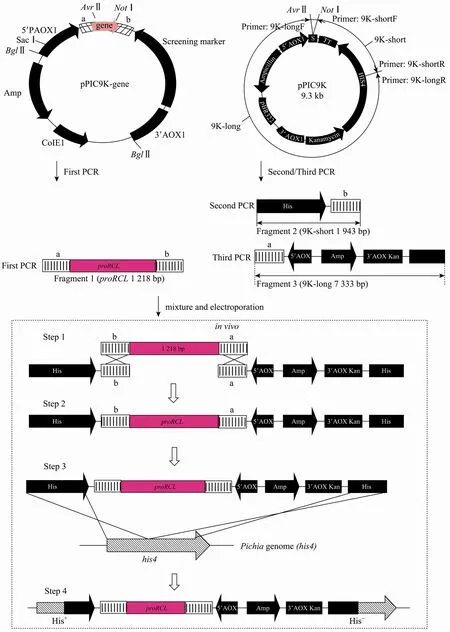
图1 基于体内同源重组构建插入型酵母突变文库的方法 (PDM) 示意图Fig. 1 Representation of the new method of mutant pool construction (mutant genes inserted into yeast genome directly) based on in vivo homologous recombination (PDM).
2.3 基因与载体同源序列长度对建库的影响
本实验最关键的步骤是基因与两段载体同源整合成为一段 DNA,而同源臂长度的选择成为同源整合能否成功的关键。本实验考察了设计不同长度同源臂对整个建库过程的影响,结果如下:
结果如表 2所示:当扩增片段和表达载体的同源臂长度为25~40 bp时,目的基因PCR扩增效果很好,但是和表达载体混合电转后,重组阳性率很低。可能是由于同源臂的长度过短,无法形成表达盒,导致脂肪酶基因不能插入到酵母基因组内。当扩增片段和表达载体的同源臂长度>70 bp时,虽然重组阳性率非常高,但是PCR扩增操作会变得比较困难,得到基因的量低,纯度差,很难满足后续实验的需要。而当同源臂长度为40~70 bp时,重组阳性率和 PCR扩增效果都具有实验的可行性,可以低成本、高效率地得到突变文库,也能保证文库筛选的效率。本实验中得到的同源臂的长度为60 bp和66 bp。
2.4 突变基因proRCL的扩增片段和pPIC9K片段的混合与电转化
基于体内同源重组构建酵母突变文库的方法见图1。将上述经3次PCR扩增得到的proRCL基因片段及9K-long和9K-short片段按照一定的摩尔比混合均匀后,电转化毕赤酵母。
进入酵母体内后,经过 4步外源基因可以同源重组进酵母基因组。
Step1:基因片段proRCL以及载体片段9K-short和9K-long末端带有的60 bp (图1中a) 及66 bp (图1中b) 重叠序列使该3段DNA发生同源重组;
Step2:3段基因连接为一段完整的DNA (即线性化后的完整的重组质粒);
Step3:线性化后的完整的重组质粒与酵母基因组同源重组;
Step4:外源基因整合进入酵母基因组。
2.5 pNPP法考察PDM法突变文库的转化率和阳性率
考察了目的基因片段与载体片段的不同摩尔比,各摩尔比结果见表3。
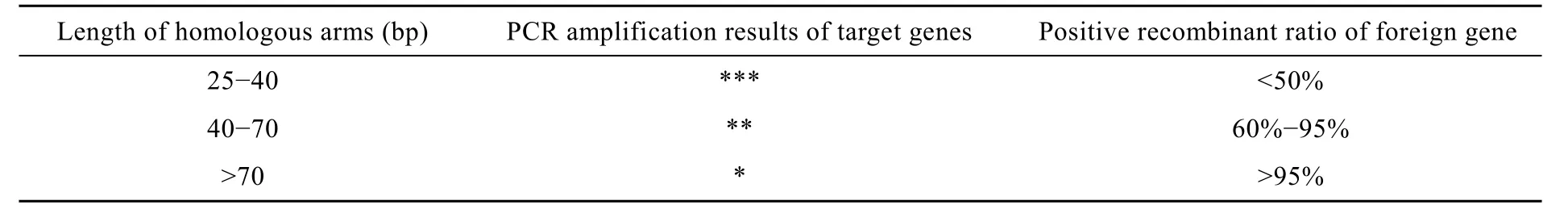
表2 同源臂长度对PCR扩增效果和重组阳性率的影响Table 2 The effective of length of homologous sequences on PCR amplification result and positive recombinant ratio

表3 转化率和阳性率表Table 3 Transformation ratio and positive recombinant ratio
如表3所示,在Error-prone PCR products:vector的比例为1∶1、3∶1、10∶1 (摩尔比) 的梯度中,载体的量为0.14 nmol、0.23 nmol和0.29 nmol。转化率变化不明显,均处在 105数量级,重组率下降明显。这是因为随着载体量的增加,未与目的基因进行连接的线性化的 9K直接与酵母基因组发生重组的概率上升。因此,1∶3和1∶10的梯度重组率更低。但是,随着载体比例增大,阳性重组率,即目的脂肪酶基因正确插入到重组酵母基因组中的概率显著下降。由此得出,最佳梯度为Error-prone PCR products∶vector=10∶1。再进行Fast-blue RR染色平板确定重组菌,其中95%都是含有目的基因的阳性菌株,具有进行后续操作的可行性。
2.6 PDM构建的重组菌的PCR鉴定
本实验最关键的步骤是基因与两段载体同源整合成为一段DNA,而基因与载体是通过同源重组在毕赤酵母体内组装成完整的、能够正确表达外源基因的表达盒,还是进行了随机整合,可以通过PCR扩增及测序来验证。以5′AOX和3′AOX为引物,对文库中目的菌株的基因组进行扩增,可以得到完整的表达盒 (图 1中左上图)。基因测序结果表明:PDM方法中目的基因与载体的确按照实验设计的方式重组整合为一段 DNA (该段DNA与亚克隆方法中构建的表达载体线性化后的片段完全相同)。
2.7 PDM 法构建的突变文库与亚克隆法构建的突变文库的比较
分别运用亚克隆方法与PDM法构建proRCL基因的突变文库,使电转化时的目的基因的摩尔数基本相同。PDM 法中 Error-prone PCR products与 vector的摩尔比为 10∶1。得到的突变文库比较如下。
由表4得,PDM法构建的基因突变文库的库容量提高了 20倍,重组阳性率提高至 95%,而建库时间不到原来的25%。
2.8 PDM法构建得到的突变菌株列表
对PDM法构建得到的突变文库进行筛选,得到如下突变菌株 (表 5),分别为酶活提高,酶表达量提高以及酶热稳定性提高的突变株。
表5中Mutant1为酶活提高的突变株,突变前后脂肪酶的表达量变化很小,但突变后脂肪酶的 Km值由原来的 0.304 mmol/(min·mg) 下降为0.258 mmol/(min·mg),下降了 15%;Kcat值由原来的 1 138 /min提高至 3 250 /min,表明了突变株Mutant1对底物的亲合力提高,比活力上升,酶的催化效率获得了提高。Mutant1是一株酶活提高的突变株。
表6中Mutant2为酶表达量提高的突变株,突变前后脂肪酶的表达量提高了3倍,Kcat值由原来的1 138 /min提高至3 520 /min,也是提高约3倍,表明 mutant2的酶活提高基本是因为表达量的提高引起的,而比活力没有提高。Mutant2是一株酶表达量提高的突变株。
表7中Mutant3为热稳定提高的突变株,突变前后脂肪酶的表达量和比活变化不大,Tm由原来的61 ℃提高至67.5 ℃,突变酶在65 ℃和70 ℃下的半衰期分别提高21倍和4.2倍,在75 ℃的半衰期也达到了7.5 min。Mutant3是一株酶热稳定性提高的突变株。

表4 亚克隆方法与PDM方法构建的突变文库比较Table 4 Comparison of mutant pools built by sub-clone method and PDM

表5 出发菌株和酶活突变菌株的动力学参数和蛋白浓度Table 5 Kinetic parameters and protein concentrations of parent strain and enzymatic mutant

表6 出发菌株和表达量突变菌株的动力学参数和蛋白浓度Table 6 Kinetic parameters and protein concentrations of parent strain and mutant of expression level

表7 出发菌株和热稳定性突变菌株的性质Table 7 Characters of parent strain and mutant of thermostability
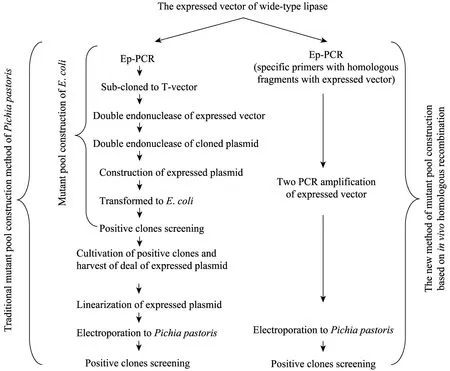
图2 亚克隆法构建酵母突变文库的方法与PDM方法的比较Fig. 2 Comparison of sub-cloned mutant pool construction method and PDM.
3 讨论
由图2得出,PDM方法效率高,操作方便:在构建得到重组表达质粒的基础上,整个建库步骤只需要一次易错 PCR、载体酶切和电转化过程,省略了传统构建过程中连接、突变质粒构建、转化大肠杆菌、提取质粒等繁琐而且耗时的工作,将建库周期由2周缩短为3 d。另外,由于该方法构建酵母基因突变文库不需要任何亚克隆步骤,降低了亚克隆步骤带来的突变基因容量与丰度的损失,库容量从传统的103~104提高到105以上。最后,该方法还具有材料与方法上的通用性,可广泛用于各种可以在酵母中获得表达的目标基因。
在酿酒酵母中Shao等利用“DNA ASSEMBLER”同源重组的方法构建了长度超过 19 kb的代谢途径[16]。本研究在毕赤酵母中成功利用体内同源重组的方法构建了快速高效的酵母整合型基因突变文库的方法。
此类方法中,决定实验成败的第1个重要因素是同源臂的长度。比如在“DNA ASSEMBLER”的方法中,随着需要同时插入基因组中基因数目的增加,同源臂的长度需随之增加,否则正确组装表达盒的概率就会下降。第2个因素是目的基因与载体PCR片段的摩尔比,摩尔比不仅与组装效率直接相关,而且还可以通过控制,来实现其他实验结果。
PDM 法是在毕赤酵母体内构建基因突变文库的非常有效的方法,但它的应用不止于此。还可以利用PDM法向毕赤酵母基因组插入代谢途径;也可以获得外源基因在毕赤酵母体内的多拷贝插入等。后者可以通过控制 PDM 中目的基因与载体的比值来实现。当目的基因与载体片段的摩尔比达到15∶1的时候,就很容易出现多基因串联形成的表达盒并插入基因组的情况,且随着摩尔比增加,表达盒中串联的基因数也随之增加。因此PDM法可高效的获得多拷贝克隆,且所需拷贝数还可以通过载体与基因的摩尔比进行控制,具体的拷贝数可以通过荧光定量的方法进行检测[17-20]。由此可见,PDM及其类似方法在利用酵母进行分子生物学研究的领域中的应用潜力非常巨大。
REFERENCES
[1] Yokoigawa K, Okubo Y, Soda K, et al. Improvement in thermostability and psychrophilicity of psychrophilic alanine racemase by site-directed mutagenesis. J Mol Catal B: Enzym, 2003, 23(216): 389−395.
[2] Chen KQ, Arnold FH. Enzyme engineering for nonaqueous solvents: random mutagenesis to enhance activity of subtilisin E in polar organic media. Biotechnology, 1991, 9(11): 1073−1077.
[3] Zhang J, Li ZQ, Zhang HY. An enzymatic method for random- (site-specific) mutagenesis of ginseng peptide gene in vitro. Chin J Biochem Mol Biol, 1992, 8(1): 115−119.
[4] Feng HY, Yi LH, Wang LL. Oligosaccharide synthesis performance of β-Glycosidase promoted by directed evolution. Chin J Bioprocess Eng, 2006, 4(1): 44−49.冯惠勇, 仪宏, 王丽丽. 定向进化技术改良 β-糖苷酶的低聚糖合成性能. 生物加工过程, 2006, 4(1): 44−49.
[5] Lee PC, Momen AZR, Mijts BN, et a1. Biosynthesis of structurally novel carotenoids in Escherichia coli. Chem Biol, 2003, 10(5): 453−462.
[6] Leong SR, Chang JCC, Ong R, et a1. Optimized expression and specific activity of IL-12 by directed molecular evolution. Proc Natl Acad Sci USA, 2003, 100(3): 1163−1168.
[7] Macauley-Patrick S, Fazenda ML, McNeil B, et al. Heterologous protein production using the Pichia pastoris expression system. Yeast, 2005, 22(4): 249−270.
[8] Scorer CA, Clare JJ, McCombie WR, et al. Rapid selection using G418 of high copy number transformants of Pichia pastoris for high level foreign gene expression. Nat Biotechnol, 1994, 12(2): 181−184.
[9] Ouyang LM, Zhang HZ, Zhang SL, et al. Advances in the studies of Pichia pastoris as a heterologous gene expression system. Prog Biochem Biophys, 2000, 27(2): 151−154.欧阳立明, 张惠展, 张嗣良, 等. 巴斯德毕赤酵母的基因表达系统研究进展. 生物化学与生物物理进展, 2000, 27(2): 151−154.
[10] Li Y, Wang D, Xu Y. Expression of the gene of pro-and mature Rhizopus chinensis lipases and associated characteristics. Biotechnol Bull, 2009(2): 82−86.李 , 王栋, 徐岩. 华根霉前导肽和成熟肽脂肪酶基因的克隆表达及性质研究. 生物技术通报, 2009(2): 82−86.
[11] Yu XW, Wang LL, Xu Y. Rhizopus chinensis lipase: gene cloning, expression in Pachia pastoris and properties. J Mol Catal B: Enzym, 2009, 57(1/4): 304−311.
[12] Wang LL, Yu XW, Xu Y. Cloning and expression of proand mature Rhizopus chinensis lipase in Pichia pastoris. Chin High Tech Letter, 2009, 19(1): 105−110.王乐乐, 喻晓蔚, 徐岩. 华根霉 (Rhizopus chinensis)前导肽脂肪酶基因的克隆及其在Pichia pastoris中的表达. 高技术通讯, 2009, 19(1): 105−110.
[13] Wu SX, Letchworth GJ. High efficiency transformation by electroporation of Pichia pastoris pretreated with lithium acetate and dithiothreitol. Biotechniques, 2004, 36(1): 152−154.
[14] Wang R, Yu XW, Sha C, et al. Enhanced enzyme activity of Rhizopus chinensis lipase by directed evolution. Chin J Biotech, 2009, 25(12): 1892−1899.王睿, 喻晓蔚, 沙冲, 等. 定向进化-易错PCR方法提高华根霉Rhizopus chinensis CCTCC M201021脂肪酶的活力. 生物工程学报, 2009, 25(12): 1892−1899.
[15] Jiang CY, Jiang T, Cao C. Study on the conditions of electrotransformation to Pichia methanolica. J Qiqihar Uni: Nat Sci, 2005, 21(1): 8−11.江成英, 江洁, 曹畅. 甲醇毕赤酵母电转化条件的研究.齐齐哈尔大学报: 自然科学版, 2005, 21(1): 8−11.
[16] Shao ZY, Zhao H, Zhao HM. DNA assembler, an in vivo genetic method for rapid construction of biochemical pathways. Nucleic Acid Res, 2009, 37(2): 1−10.
[17] Hartner FS, Ruth C, Langenegger D, et al. Promoter library designed for fine-tuned gene expression in Pichia pastoris. Nucleic Acids Res, 2008, 36(12): e76.
[18] Schroer K, Luef KP, Hartner FS, et al. Engineering the Pichia pastoris methanol oxidation pathway for improved NADH regeneration during whole-cell biotransformation. Metab Eng, 2010, 12(1): 8−17.
[19] Williams KE, Jiang J, Ju JL, et al. Novel strategies for increased copy number and expression of recombinant human gelatin in Pichia pastoris with two antibiotic markers. Enzyme Microb Tech, 2008, 43(1): 31−34.
猜你喜欢
华人时刊(2023年1期)2023-03-14
汉字汉语研究(2021年2期)2021-08-30
猪业科学(2021年3期)2021-05-21
幽默大师(2020年10期)2020-11-10
中华诗词(2019年1期)2019-11-14
汉字汉语研究(2019年2期)2019-08-27
猪业科学(2018年4期)2018-05-19
国外医药(抗生素分册)(2016年2期)2016-07-12
河北书画研究(2016年3期)2016-04-28
中国塑料(2016年7期)2016-04-16
