
PepT1靶向前药的研究进展
2011-02-02 06:17平其能
药学进展 2011年1期
印 志, 操 锋, 平其能
(中国药科大学药剂学教研室,江苏南京 210009)
PepT1靶向前药的研究进展
印 志, 操 锋, 平其能*
(中国药科大学药剂学教研室,江苏南京 210009)
PepT1是一种主要分布于小肠的寡肽转运器,对食物中蛋白质水解所得的二肽和三肽的胃肠道吸收发挥着重要作用。通过化学修饰,使一些膜通透性差的药物对PepT1具有一定的亲和力,能经PepT1转运透过细胞膜,以改善药物的膜通透性,达到提高药物口服生物利用度的目的,这一策略已成为当前口服靶向前药研究领域中的热点。综述近年来PepT1靶向前药的研究进展,包括PepT1的转运机制、PepT1底物的模型以及不同结构类别的该转运器靶向前药实例。
靶向前药;寡肽转运器1;转运机制;酯类前药
越来越多的研究表明,在很多药物的小肠吸收过程中,跨膜转运蛋白参与的主动转运起着关键的作用。而且,这些转运蛋白不仅分布于小肠,还分布于肝、肾、脑等部位,广泛参与药物的吸收、分布、代谢及排泄,最终影响药物的疗效和毒性大小[1]。
PepT是一种质子依赖型的转运蛋白,具有12个跨膜区域和膜外的糖基化结构。目前所知的寡肽转运蛋白PepT主要位于上皮细胞的刷状缘膜和基底外侧膜[2]。位于刷状缘膜的PepT有两种常见亚型,即PepT1和PepT2;而基底外侧膜处PepT是否亦有不同的亚型目前尚不明了。PepT1主要分布于小肠上皮细胞,主要转运食物中蛋白质水解的二肽和三肽,也参与一些口服使用的药物,如β-内酰胺类抗生素头孢羟氨苄和阿莫西林、血管紧张素转化酶抑制剂福辛普利和依那普利、抗病毒药物贝他定和伐昔洛韦等的吸收[3]。另外还有研究发现,在眼部血液-房水屏障和血液-视网膜屏障、肝外胆管上皮细胞顶膜、肾脏近端小管处亦有少量的PepT1表达[4-6];而PepT2则主要分布于肾,对由肾小球滤入血液中的寡肽进行重吸收,另在脉络丛及肺部等处也有分布[7-8]。
PepT1属于低亲和力,高转运能力的转运蛋白,可以识别广泛范围内的不同大小、电荷的药物分子。另外由于这种主动转运系统的Km和Vmax都比较大,肠道高浓度药物不易使其转运能力达到饱和,这些特性使得靶向PepT1的研究成为口服靶向前药中最热门的领域。研究人员试图通过化学修饰,将那些膜通透能力差的药物制成对PepT1具有亲和力的前药,以便借助PepT1将其转运进入肠细胞,从而达到提高药物的口服生物利用度的目的。而且因前药改变了母体药物的分子构型,还有可能避开母体药物所受外排转运器的外排作用,进一步增加药物的体内有效浓度。本文就PepT1的转运机制、PepT1底物的特征以及各类典型的靶向PepT1的前药加以介绍。
1 PepT1的转运机制
PepT1转运过程分为三个步骤(如图1):首先是小肠上皮细胞基底外侧膜的Na+/K+-ATPase通过Na+/K+交换,向细胞外泵出Na+,产生细胞内外Na+浓度梯度;而后,这种梯度驱动位于细胞顶膜侧的Na+/H+交换转运器(NHE3)将H+转运至细胞外,随着细胞外侧的H+浓度上升,细胞内外产生H+浓度梯度差和负的膜电位;最后,PepT1在这两种梯度的协同作用下,将药物或寡肽转运进入细胞内[9]。

图1 小肠PepT1的转运机制Figure 1 Transportmechanism of PepT1 in intestine
依据PepT1转运机制,PepT1的转运是一个质子驱动的过程,肠道中的pH环境对底物的吸收起着关键作用。因此,尽管从十二指肠至回肠段的PepT1表达逐渐增多,但是由于该段环境中的pH也逐步升高,故在正常的生理条件下,PepT1底物的转运并不能得到相应增加[10],而若适当降低肠道的pH,将有利于PepT1对其底物的转运。如抗菌药头孢克肟本身就是PepT1的底物,需经PepT1主动转运而被吸收,Nozawa等[11]在头孢克肟中添加酸性辅料Eudragit L100-55后给大鼠灌胃(用量以头孢克肟2.26 mg/kg计),结果发现,这种添加了酸性辅料的头孢克肟的生物利用度是采用普通辅料的头孢克肟的2.3倍。该研究提示,如果在将药物制成PepT1靶向前药的同时,再结合添加适当的酸性辅料,便可以充分发挥PepT1的转运能力,协同提高药物的口服生物利用度。
2 PepT1靶向前药的底物模型
PepT1靶向前药是指以 PepT1为靶标,利用PepT1的主动转运来增加药物膜透过性,以提高生物利用度的前药。因此,充分、全面地了解PepT1的结构对于合理设计PepT1靶向前药具有重要的意义。目前虽然已对PepT1的大致结构、功能和分布等有比较清楚的了解,但是由于尚未获得这类跨膜蛋白的结晶,所以还不知晓PepT1完整的3D结构及其与底物的结合位点,故人们只能通过研究已知底物的3D结构来预测PepT1底物的结构要素[12]。例如Bailey等[13]研究了100多个PepT1底物的结构特征,总结出PepT1底物的结构特性包括:1)一个能与氨基端NH3+结合的位点;2)氨基端α-C具有一定的立体化学选择性(通常是L-型);3)氨基端α-C与R2之间有一段展开的平面长链;4)与第一个肽键的羰基形成氢键;5)N2可以被烷基化;6)第二个残基处的三个基团有特定取向(通常是L-型); 7)一个有立体选择性的疏水性口袋;8)一个羧酸盐结合位点;9)足够容纳侧链R3的空间;10)确定临近手性中心取向的第二个羧酸盐结合位点(通常是L-型)(参见图2)。

图2 PepT1与底物的结合及底物的结构特征Figure 2 Substrates binding to PepT1 and their structural characteristics
另外,一些已知的底物,如对氨基苯乙酸、δ-氨基乙酰丙酸以及ω-氨基脂肪酸等,结构中并没有肽键,但是仍然对PepT1有较高的亲和力,这说明肽键在底物对PepT1的识别中可能并不是必须的[2]。
目前PepT1靶向前药的主要设计思路是用具有特定空间结构的氨基酸或寡肽修饰母体药物,使其与PepT1有较强的亲和力。
3 PepT1靶向前药的设计
3.1 PepT1靶向酰胺类前药
将一些药物原有的游离氨基与配体氨基酸或短肽的羧基形成肽键,即可形成与PepT1亲和力较强的酰胺类的前药。如礼来公司开发了一种抗焦虑药物LY354740(1),其经注射使用时,对多种动物实验模型均有较好的疗效,但由于其水溶性高,生物透过能力差,属于生物药剂学分类系统(Biopharmaceutical Classification System,BCS)中第三类,故在给大鼠和狗经口使用后,体内生物利用度仅分别为10%和45%。为此,研究人员将LY354740的氨基与丙氨酸以肽键相连,得到LY544344(2),在体肠灌流实验结果显示,当 LY544344的浓度为0.1 mmol·L-1时,渗透系数 Peff比 LY354740 提高了10倍左右,且除约10%通过被动扩散进入细胞外,大部分LY544344均是经由PepT1转运进入细胞,并在细胞内被包括金属肽酶在内的多种肽酶几乎完全水解成 LY354740,再通过被动扩散进入血液[14-16]。
2.1.2 原因分析(见表2) 由表2可以看出,影响医学生对思想政治教育感兴趣的因素主要有:(1)学校思想政治教育的形式。有32.7%的医学生认为思想政治教育形式单一,基本以理论教学为主,太枯燥。(2)医学生对思想政治教育的认识水平。有17.7%的医学生认为思想政治教育不重要,学好专业课才是根本。(3)学习负担过重。11.3%的医学生认为学习负担过重,无暇顾及思想政治学习,在有限的时间内,首当其冲先学习专业课程。

再如,用于治疗体位性低血压的选择性α1-受体激动剂甲氧胺福林(midodrine,3)是在脱甘氨酸米多君(DMAE,4)分子支链的氨基上结合了甘氨酸后形成的酰胺类前药,其真正有效的成分为DMAE。甲氧胺福林由PepT1转运入体内,口服生物利用度由DMAE的50%提高至93%[17],进入体内后,再在肝脏和血浆中被某种肽酶水解成母体药物DMAE而发挥疗效。

3.2 PepT1靶向酯类前药
氨基酸的羧基与药物的氨基偶联形成的酰胺类前药可能存在引入的酰胺键在体内过于稳定、无法水解成具有药理活性的母体药物的问题,因此目前报道的PepT1靶向酰胺类前药仅有两个,而更多的是将药物的游离羟基与氨基酸的羧基酯化后形成的酯类前药。
3.2.1 经典PepT1靶向酯类前药
抗病毒药物伐昔洛韦(Valacyclovir,LACV,5)是目前已上市的最成功的PepT1靶向前药。其母体药物阿昔洛韦(Acyclovir,ACV)的水溶性差,难以透过

3.2.2 减少P-糖蛋白外排的PepT1靶向前药
P-糖蛋白(P-Glycoprotein,P-gp)是一种 ATP 依赖泵,作为一种转运蛋白在正常组织中广泛表达,并且配体多样,其生理功能可影响到药物代谢的全过程。例如小肠上皮细胞中的P-糖蛋白,可将进入小肠细胞内的药物外排至肠腔,导致很多药物的口服生物利用度下降。虽然采用P-gp抑制剂或者运用pluronic P85等材料将药物包裹在胶束中,可以避免P-gp对药物的外排作用[21],但是这些方法存在一定的安全性问题,因此有研究者试图通过直接改变药物的结构来避开P-gp的外排,并且,这些结构修饰兼顾了药物对PepT1的靶向亲和作用,故有助于进一步提高其口服生物利用度。
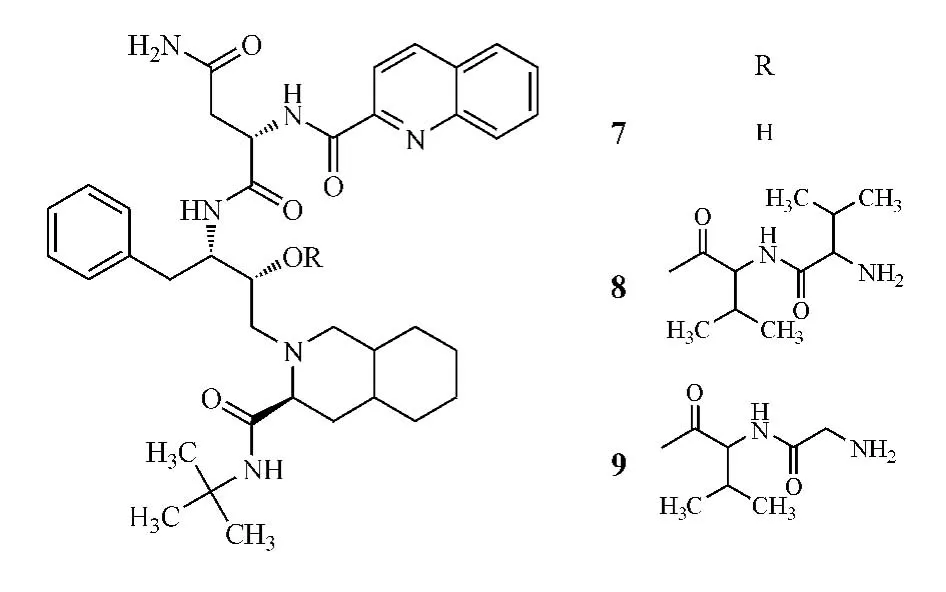
研究表明,绝大部分抗HIV蛋白酶抑制剂均是P-gp的底物,并且这些药物还是肝和消化道CYP3A4酶的底物,因此这些药物易于被大量降解或外排,导致口服生物利用度下降,如第一个获得FDA批准的、用于治疗艾滋病的HIV蛋白酶抑制剂沙奎那韦(saquinavir,SQV,7)便是如此。Jain 等[22]用二肽缬氨酸-缬氨酸-(Val-Val-)和甘氨酸-缬氨酸肠道上皮细胞,口服生物利用度只有20%。而伐昔洛韦是用L-缬氨酸将阿昔洛韦的3-OH酯化得到的前药,其为PepT1的底物,经小肠上皮的PepT1转运进入体内,再经肠道和肝脏首过作用转化为母体药物阿昔洛韦发挥抗病毒活性。这一结构的修饰使得伐昔洛韦的口服生物利用度比阿昔洛韦提高了3~5 倍[18-19]。
另一个酯类前药的例子是抗病毒药物更昔洛韦(ganciclovir)的 L-缬氨酸酯——缬更昔洛韦(Valganciclovir,6)。缬更昔洛韦比母体药物更昔洛韦对PepT1有更高的亲和力,能通过PepT1转运进入体内,再经小肠细胞的酯酶水解成更昔洛韦发挥药效。更昔洛韦的口服生物利用度仅为6%,而缬更昔洛韦的口服生物利用度则提高至61%[20]。-(Gly-Val-)将沙奎那韦的羟基酯化得到了 Val-Val-SQV(8)和 Gly-Val-SQV(9),大鼠肠道灌流实验显示,两种前药的空肠吸收速率常数分别为SQV的4.6和1.8倍。该作者认为,这与两种前药进入消化道与PepT1结合后发生空间构象的改变,导致P-gp对其无法识别有关。
3.2.3 眼用PepT1靶向前药
虽然在某些情况下,口服或静脉注射给药也是治疗眼部疾病的有效给药途径,但是药物经这些途径进入体内后均需透过血-眼屏障(blood ocular barriers,BOB)才能到达眼睛内部组织,发挥疗效,而因BOB由血液-房水屏障和血液-视网膜屏障组成,一些水溶性的药物分子,如阿昔洛韦(ACV)和更昔洛韦在口服或静注给药后无法透过BOB。但由于在BOB上也存在PepT,因此通过对此类药物的结构修饰,将其制成合适的前药形式,使其能利用PepT转运透过BOB,以提高药物的眼内生物利用度是一个可行的办法。但难点在于必须保证前药在系统循环中保持良好的稳定性,不会被系统循环中的酶(主要是酯酶或者肽酶)水解,能以前药的形式到达BOB处,并与此处的PepT结合进入眼内部后方才水解出母体药物。Talluri等[23]根据水解酶具有立体特异性、且对L-异构体的亲和力较强的特点,采用氨基酸的D-异构体作为修饰物制备前药,使其在循环系统中不易被酶水解。他们对分别用D-缬氨酸-L-缬氨酸、L-缬氨酸-L-缬氨酸、L-缬氨酸-D-缬氨酸和 D-缬氨酸-D-缬氨酸修饰的 ACV:DLACV、LLACV、LDACV 和 DDACV 在 Caco-2细胞匀浆、大鼠肠匀浆、肝匀浆中的稳定性进行了比较。结果表明,这些前药的稳定性大小顺序为:DDACV >DLACV>LDACV>LLACV。另外研究者以AVC及其经典PepT1靶向前药伐昔洛韦(LACV)为参照,考察了前药在Caco-2细胞的透过性。结果显示,除DLACV外,其余前药在Caco-2细胞的透过性均高于ACV,其中LDACV的表观透过率Papp最高,是LACV的2倍(如图3)。

图3 前药在Caco-2表观透过率(cm·s-1)Figure 3 Apparent permeability of all the prodrugs across Caco-2
上述结果说明:仅掺入一个D-缬氨酸的前药可保持对PepT的亲和力,且能有效地降低水解酶的水解,更好地实现对血-眼屏障处PepT的靶向作用。另外,部分药物可通过局部给药途径,用于治疗眼前房的病变。但有人研究发现,在人类和家兔的角膜上都存在着外排泵 P-gp[24],很多眼用药物,如甾体或非甾体类抗炎药、β-受体阻滞剂、抗生素等等均是P-gp的底物,可被 P-gp外排至角膜前液,导致局部使用药物的眼部生物利用度下降。因此,利用角膜上皮上表达的PepT1提高这些药物的生物利用度成为了人们的研究课题。如Katragadda等[25]将典型的P-gp抑制剂——奎尼丁分别用 L-缬氨酸和 L-缬氨酸-缬氨酸进行酯化,得到了VQ和VVQ,结果,两种酯化前药在兔角膜的透过率分别比奎尼丁提高了1.5倍和3倍;且兔角膜细胞摄取实验发现,当有PepT1的经典底物甘氨酰肌氨酸(glycyl-sarcosins,Gly-Sar)存在时,VQ和VVQ的转运明显下降,说明这两种前药角膜透过率的提高与其和PepT1的亲和力及竞争性结合有关;另用[14C]标记的红霉素进行的实验表明,当有奎尼丁存在时,由于P-gp被抑制,作为其底物的红霉素的吸收增加了71%;而若在VQ和VVQ存在的情况下,[14C]红霉素的吸收并无明显增加,说明这两种前药对P-gp的作用比其母体药物奎尼丁小得多。
3.3 巯基二肽载体前药

目前大多数PepT1靶向前药的设计思路是模拟PepT1的天然底物,将生物药剂学性质差的药物直接修饰成类似于二肽或三肽的前药。但是这类前药几乎都是依赖酯键或者酰胺键与母体药物相连,这意味着候选药物必须含有羟基、氨基或者羧基,因此使得这种方法的使用范围受到了限制。Bailey等[26]根据 PepT1 天然底物 Phe-Ala(10)设计了巯基二肽载体(11),其选用合适的连接臂将载体与酮类药物相连,将氧替换成硫后不仅提高了跨膜转运速率,而且能有效避免肽键被水解,提高了该类药物对PepT1的靶向性。利用这种载体可以将更多种类的药物修饰成PepT1靶向前药,解决药物的膜透过性难题。研究人员称之为“巯基二肽载体前药手段(thiodipeptide“carrier”prodrug approach)”。还有研究者选择非甾体抗炎药萘丁美酮(12)作为模型药物,考察上述载体能否提高药物对PepT1靶向性。虽然酮类药物的羰基不可以直接与连接基团相连,但研究表明,肟类化合物能够在体内被细胞色素P450水解成酮类,所以他们先将萘丁美酮成肟(13),通过特定链长的聚乙二醇与巯基二肽相连得到两个萘丁美酮的前药(14,15)。用Caco-2细胞考察这两个萘丁美酮前药的靶向性,结果表明,当聚乙二醇重复单元数n为0和2时,两个前药的表观渗透性系数Papp分别是一种 PepT1的典型底物——Pheψ[CS-NH]-Ala(FSA)的1.3 倍和6.3 倍。说明萘丁美酮的巯基二肽前药能利用PepT1跨膜转运,迅速进入上皮细胞内部,这对于改善药物的生物利用度是非常有利的[27]。另外,Foley 等[28]将苯甲醇和苯甲酸与不同嵌段长度的乙二醇以醚键或酯键相连,再连接巯基二肽,得到一系列不同链长的巯基二肽载体前药。结果显示当乙二醇嵌段以单一酯键与载体巯基二肽相连时,连接臂越长,PepT1介导的化合物转运速率越慢。
这种以巯基二肽为载体提高药物PepT1靶向性的方法无疑为研究者拓宽了思路,更多结构的药物可以被合理巧妙地修饰成PepT1靶向前药,以改善自身的膜透过性问题,提高口服生物利用度。

4 结语
目前,制备PepT1靶向前药已成为提高和改善某些水溶性不佳、膜通透性差药物的口服生物利用度的有效手段之一。而用人体必需氨基酸或者短肽对母体药物进行结构修饰,以形成前药,除可利用前药与PepT1的亲和力达到促进吸收的目的之外,因其进入人体后既能释放出母体药物,也能释放人体必需的氨基酸,故还可能有一定的营养作用。
但是,至今为止,可供修饰的母体药物在结构上仍受到较大的限制:只有分子结构中含有羟基、羧基、氨基或者羰基等官能团的药物才有可能被制成上述前药。因此,目前需要进行深入探讨的课题之一是如何能将具有不同分子结构或基团的药物改造成对PepT1有亲和力、并能在进入体内后分解和释放出母体药物的前药。
另一个需要解决的问题是如何保证前药能在适当的时间和适当的部位分解和释放,因为所制得的前药必须有合适的酶稳定性:药物既不能在被PepT1转运之前降解,也不能在进入血液后迟迟不降解而仍以前药形式存在,否则均无法发挥药效。
此外,由于目前对这类前药的渗透性和靶向性研究多以Caco-2细胞为试验模型,而有研究表明,Caco-2细胞表达PepT1的水平低于体内的相关细胞,因此选择出高表达PepT1的细胞模型以便能在体外优选出靶向效果好的前药也成为了PepT1靶向前药研究过程中的热点课题,现已有人在试验使用PepT1 转染的各种细胞模型,如 HeLa/hPepT1[29],CHO/hPepT1[30]或 MDCK/hPepT1[31]细胞模型等。
再有,大量研究已表明PepT1表达和活性受多种因素,如底物、激素、生长因子、饮食条件、生长发育、昼夜节律等的调节[2],如 Ihara 等[32]研究发现,短期的饥饿可以使转运器表达显著上调,而饮食摄入氨基酸会降低肠道中PepT1的水平。这些因素可能会对PepT1靶向前药的体内药动学以及细胞内的信号传递产生较大的影响。故如何利用这些生理因素以及采用特殊的生物分子等诱导转运器表达的上调、进一步促进这类靶向药物的体内吸收也是非常值得研究的课题。
[1]Dobson PD,Kell D B.Carrier-mediated cellular uptake of pharmaceutical drugs:an exception or the rule[J].Nat RevDrugDiscov,2008,7(3):205-220.
[2]Terada T,Inui K.Peptide transporters:structure,function,regulation and application for drug delivery[J].CurrDrug Metab,2004,5(1):85-94.
[3]Meredith D.The mammalian proton-coupled peptide cotransporter PepT1:sitting on the transporter-channel fence? [J].PhilosTransRSocLondBBiolSci,2009,364(1514):203-207.
[4]Dias C,Nashed Y,Atluri H,etal.Ocular penetration of acyclovir and its peptide prodrugs valacyclovir and valvalacyclovir following systemic administration in rabbits: An evaluation using ocularmicrodialysis and LC-MS[J].CurrEyeRes,2002,25(4):243-252.
[5]Atluri H,Anand B S,Patel J,etal.Mechanism of amodel dipeptide transport across blood-ocular barriers following systemic administration[J].ExpEyeRes,2004,78(4): 815-822.
[6]Lu H,Klaassen C.Tissue distribution and thyroid hormone regulation of PepT1 and Pept2 mRNA in rodents[J].Peptides,2006,27(4):850-857.
[7]Teuscher N S,Novotny A,Keep R F,etal.Functional evidence for presence of PepT2 in rat choroid plexus: studies with glycylsarcosine[J].JPharmacolExpTher,2000,294(2):494-499.
[8]Groneberg D A,Nickolaus M,Springer J,etal.Localization of the peptide transporter PepT2 in the lung:implications for pulmonary oligopeptide uptake[J].AmJPathol,2001,158(2):707-714.
[9]Brandsch M.Transport of drugs by proton-coupled peptide transporters:pearls and pitfalls[J].ExpertOpinDrug MetabToxicol,2009,5(8):887-905.
[10] Naruhashi Kazumasa,SaiYoshimichi,Tamai Ikumi,etal. PepT1 mRNA expression is induced by starvation and its level correlates with absorptive transport of cefadroxil longitudinally in the rat intestine[J].PharmRes,2002,19 (10):1417-1423.
[11] Nozawa Takashi,Toyobuku Hidekazu,KobayashiDaisuke,etal.Enhanced intestinal absorption of drugs by activation of peptide transporter PepT1 using proton-releasing polymer[J].JPharmSci,2003,92(11):2208-2216.
[12] Meredith D,Price R A.Molecularmodeling of PepT1 —towards a structure[J].JMembrBiol,2006,213(2):79-88.
[13] Bailey P D,Boyd C A,Bronk JR,etal.How to Make drugs orally active:a substrate template for peptide transporter PepT1[J].AngewChemIntEdEngl,2000,39 (3):505-508.
[14] Eriksson A H,Varma M V,Perkins E J,etal.The intestinal absorption of a prodrug of themGlu2/3 receptor agonist LY354740 ismediated by PepT1:in situ rat intestinal perfusion studies[J].JPharmSci,2010,99(3): 1574-1581.
[15] Varma M V,Eriksson A H,Sawada G,etal.Transepithelial transport of the group II metabotropic glutamate 2/3 receptor agonist(1S,2S,5R,6S)-2-aminobicyclo[3.1. 0]hexane-2,6-dicarboxylate(LY354740)and its prodrug (1S,2S,5R,6S)-2-[(2'S)-(2'-amino)propionyl]aminobicyclo[3.1.0]hexane-2,6-dicarboxylate(LY544344)[J].DrugMetabDispos,2009,37(1):211-220.
[16] Perkins E J,Abraham T.Pharmacokinetics,metabolism,and excretion of the intestinal peptide transporter 1 (SLC15A1)-targeted prodrug(1S,2S,5R,6S)-2-[(2' S)-(2-amino)propionyl]aminobicyclo[3.1.0]hexen-2,6-dicer- boxylic acid(LY544344)in rats and dogs: assessment of first-pass bioactivation and dose linearity[J].DrugMetabDispos,2007,35(10):1903-1909.
[17] Tsuda Masahiro,Terada Tomohiro,Irie Megumi,etal. Transport characteristics of a novel peptide transporter 1 substrate,antihypotensive drug midodrine,and its amino acid derivatives[J].JPharmacolExpTher,2006,318 (1):455-460.
[18] Guo A,Hu P,Balimane P V,etal.Interactions of a nonpeptidic drug,valacyclovir,with the human intestinal peptide transporter(hPepT1)expressed in amammalian cell line[J].JPharmacolExpTher,1999,289(1):448-454.
[19] Beutner K R.Valacyclovir:a review of its antiviralactivity,pharmacokinetic properties,and clinical efficacy[J].AntiviralRes,1995,28(4):281-290.
[20] Sugawara M,HuangW,Fei Y J,etal.Transportof valganciclovir,a ganciclovir prodrug,via peptide transporters PepT1 and PepT2[J].JPharmSci,2000,89(6): 781-789.
[21] Shaik N,Giri N,ElmquistW F.Investigation of themicellar effect of pluronic P85 on P-glycoprotein inhibition:cell accumulation and equilibrium dialysis studies[J].J PharmSci.2009,98(11):4170-4190.
[22] Jain R,Duvvuri S,Kansara V,etal.Intestinal absorption of novel-dipeptide prodrugs of saquinavir in rats[J].IntJ Pharm,2007,336(2):233-240.
[23] Talluri R S,Samanta S K,Gaudana R,etal.Synthesis,metabolism and cellular permeability of enzymatically stable dipeptide prodrugs of acyclovir[J].IntJPharm,2008,361(1-2):118-124.
[24] Dey S,Patel J,Anand B S,etal.Molecular evidence and functional expression of P-glycoprotein (MDR1) in human and rabbit cornea and corneal epithelial cell lines[J].InvestOphthalmolVisSci,2003,44(7):2909-2918.
[25] Katragadda S,Talluri R S,Mitra A K.Modulation of P-glycoprotein-mediated efflux by prodrug derivatization:an approach involving peptide transporter-mediated influx across rabbit cornea[J].JOculPharmacolTher,2006,22 (2):110-120.
[26] Bailey P D.Drug delivery system comprising a thiopeptide,EP,WO2005067978[P].2005-07-28.
[27] Foley D,Bailey P,Pieri M,etal.Targeting ketone drugs towards transport by the intestinal peptide transporter,PepT1[J].OrgBiomolChem,2009,7(6):1064-1067.
[28] Foley D,PieriM,Pettecrew R,etal.The in vitro transport of model thiodipeptide prodrugs designed to target the intestinal oligopeptide transporter,PepT1[J].OrgBiomol Chem,2009,7(18):3652-3656.
[29] Sun J,Dahan A,Amidon G L.Enhancing the intestinal absorption of molecules containing the polar guanidino functionality:a double-targeted prodrug approach[J].JMedChem,2010,53(2):624-632.
[30] Fujisawa Yuki,Tateoka Ryoko,Nara Toshifumi,etal.The extracellular pH dependency of transport activity by human oligopeptide transporter 1(hPEPT1)expressed stably in Chinese hamster ovary(CHO)cells:a reason for the bell-shaped activity versus pH[J].BiolPharm Bull,2006,29(5):997-1005.
[31] Tsume Yasuhiro,Vig B S,Sun J,etal.Enhanced absorption and growth inhibition with amino acid monoester prodrugs of floxuridine by targeting hPEPT1 transporters[J].Molecules,2008,13(7):1441-1454.
[32] Ihara Takashi,Tsujikawa Tomoyuki,Fujiyama Yoshihide,etal.Regulation of PepT1 peptide transporter expression in the rat small intestine under malnourished conditions[J].Digestion,2000,61(1):59-67.
(责任编辑:孙大柠)
Advances in Research on Prodrug Targeted to PepT1
YIN Zhi, CAO Feng, PING Qi-neng
(DepartmentofPharmaceutics,ChinaPharmaceuticalUniversity,Nanjing210009,China)
Peptide Transporter 1(PepT1),which is predominantly distributed throughout the small intestine,plays an important role in the absorption of di-and tripe ptides from the digestion of ingested protein.A lot of poorly absorbed drugs have been modified into prodrugs by affinity for PepT1 to improve permeability across intestinal membrane,and eventually to enhance the oral bioavail ability of the drugs. Recently,this strategy has become a hot topic of research on targeted prodrug.This review has addressed progress of the prodrugs targeted to PepT1 in recent years,including transport mechanism of PepT1,substrate templet for binding to PepT1,and classic PepT1-targeted prodrugs with different structures.
targeted prodrug;Peptide Transporter 1;transportmechanism;ester prodrug
R 914.2
A
1001-5094(2011)01-0015-08
10.3969/j.issn.1001-5094.2011.01.003
[接受日期]2010-09-01
[项目资助]国家自然科学基金(No.81001413);教育部新教师基金(No.20090096120002);国家重大新药创制科技重大专项(No.2009ZX09310-004)
*通讯作者:平其能,教授;
研究方向:药物新剂型和新技术;
Tel:025-83271079; E-mail:pingqn2004@yahoo.com.cn
猜你喜欢
中老年保健(2022年1期)2022-08-17
肝博士(2022年3期)2022-06-30
保健医苑(2022年5期)2022-06-10
中国典型病例大全(2022年12期)2022-05-13
中国典型病例大全(2022年7期)2022-04-22
中国药学药品知识仓库(2022年5期)2022-04-11
皮肤病与性病(2021年3期)2021-07-30
昆明医科大学学报(2021年2期)2021-03-29
教育周报·教育论坛(2020年3期)2020-10-21
科技资讯(2018年16期)2018-10-26
