
DNA聚合酶Ⅰ的功能与结构的发现
2011-01-24 08:04向义和
自然杂志 2011年6期
向义和
教授,清华大学物理系,北京 100084
本文具体介绍了DNA聚合酶Ⅰ的功能与结构的发现过程,包括聚合酶活性中心模型的提出;聚合酶分子量的测定;聚合酶的分离及其活性片段的获得;聚合酶Klenow片段晶体结构的测定以及DNA结合部位与活性位点的确立;Klenow片段结合DNA的晶体结构以及噬菌体T7DNA复制的晶体结构的测定,在测定这些复合物共晶体的基础上,提出了DNA与聚合酶结合的右手模型。
大肠杆菌DNA聚合酶Ⅰ(DNA polymeraseⅠ,缩写为polⅠ)是在1957年由科恩伯格(A.Kornberg)及其合作者发现的第一个DNA聚合酶。在各种DNA聚合酶中,这种酶研究得最透彻。它的一些特点代表了DNA聚合酶的基本特点,显示出DNA聚合酶的主要特征。DNA聚合酶Ⅰ是一种多功能酶,除了具有5′→3′聚合酶活性以外,还具有3′→5′外切核酸酶活性和5′→3′外切核酸酶活性。
1969年,科恩伯格研究了各种各样的DNA结构与聚合酶的结合,提出了DNA聚合酶的活性中心模型,显示了在这个活性中心内的几种活性位点,表示了错配碱基被外切核酸酶切除和DNA聚合酶的修复过程。同年,科恩伯格和他在斯坦福大学医学院生物化学系的同事还用沉降平衡法确定了DNA聚合酶的相对分子量,并通过蛋白水解作用把DNA聚合酶剪切成两个片段,较大的片段保持了聚合酶活性和3′→5′外切核酸酶活性,小片段包含了5′→3′外切核酸酶活性。1985年,Steitz等人获得了第一个聚合酶的晶体结构,使人们首次看到DNA合成机制的细致结构。他们提出了DNA结合部位和3′→5′外切核酸酶活性位置。1993年,他们又获得了DNA聚合酶Ⅰ的大片段——Klenow片段结合DNA的晶体结构,提出了一个在聚合酶裂缝中,DNA的结合位置模型。1998年,Doublie等人获得了噬菌体T7DNA复制的晶体结构,以2.2Å最短距离分辨的复合物。2008年,沃森(J.D.Watson)等人根据Doublie等人的实验结果描绘出DNA与聚合酶结合的右手模型图。
1 聚合酶活性中心模型的提出
1969年,科恩伯格把研究DNA聚合酶功能的成果总结成题为“DNA聚合酶的活性中心”的论文,发表在Science第163卷上。文章一开始就写道:“现在已经从各种各样细菌的和动物的细胞中分离出DNA聚合酶,包括随病毒感染而特别产生的这些酶,它们起催化作用,把单核苷酸单位加到引物DNA链的3′-羟基端。因此合成在5′→3′方向进行。绝对需要有一个DNA模板,而且在复制模板时很少发生错误。DNA的合成以每个酶分子每分钟大约加上1000个核苷酸的速率迅速地进行,随之产生具有几百万分子量的链。”[1]
科恩伯格小组通过沉降平衡确定了均匀聚合酶的相对分子量是109 kDa。这个大分子量以及聚合酶和多种核酸酶活性的存在表明,DNA聚合酶具有亚单位结构。他们做了聚丙烯酰胺凝胶电泳实验。实验表明,超过95%的蛋白质按照聚丙烯酰胺凝胶电泳迁移到pH值为3.5,8和11处形成三条单一的带。他们认为:“这个结果表明DNA聚合酶是与由单条多肽链组成的结构,或者是由两个或更多个相同的亚单位组成的结构。然而,由于每个分子量是109 kDa的聚合酶包含一个硫氢基(SH)和一个二硫键(在两个硫原子之间形成的共价键)基,多个相同亚单位组成的可能性就被排除在外。因为用碘乙酸或者用汞离子能够改变硫氢基,从而产生具有全部聚合酶和外切核酸酶活性的衍生物,所以这个硫氢基大概不是活性位置的部位。这个带有汞离子的反应或是产生每个蛋白质分子具有一个汞原子的聚合酶单体,或是在存在过量酶的情形下产生具有由汞原子连接的两个蛋白质分子的二聚体。这个二聚体也有全部活性。”[1]
1.1 DNA结合部位
他们用各种各样的DNA结构(图1)研究DNA与聚合酶的结合。用脱氧核糖核酸酶部分地水解脱氧腺苷酸和脱氧胸苷酸交替的共聚物(dAT),通过凝胶过滤或聚丙烯酰胺凝胶电泳从这些水解物中分离出寡聚体,获得了大约具有24个和40个核苷酸残基(d(AT)12和d(AT)20)链长的制剂。根据寡聚体在低离子浓度中的熔解和迅速地冷却导致他们假设这个寡聚体具有发夹型结构。
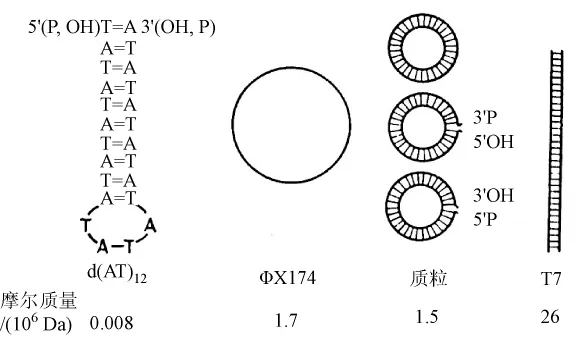
图1 在研究酶结合中使用的各种各样的DNA结构
他们用包含有3H或32P标记的DNA和203Hg标记的聚合酶的混合物的蔗糖密度梯度离心测量DNA与聚合酶的结合。这个混合物在梯度的顶端分层,在沉降后辨别DNA与酶的复合物。用过量的酶,d(AT)12与酶几乎定量地沉降,说明它们之间具有很高的结合亲和力。自由酶以沉降系数6.1S沉降,而这个聚合酶-dAT复合物是以7.7S沉降。具有过量存在的dAT寡聚体,所有的酶像一个具有等摩尔量dAT的复合物以7.7S沉降,说明DNA聚合酶包含一个与dAT寡聚体结合的位置。这些结果与用d(AT)20获得的结果一样。在把末端是3′-羟基和3′-磷酸基的寡聚体的结合加以比较时,没有检测到差别。
聚合酶与单链环形ΦX174DNA结合,在这个复合物中每个DNA分子大约结合20个酶分子。一个双链闭合环状质粒DNA完全不与聚合酶相结合。然而,当使这些双链形式变性成为单链时,单链以正比于它的长度与聚合酶结合,而且达到像单链病毒ΦX174DNA相同的程度。
他们已经用胰性脱氧核糖核酸酶把切口引入质粒DNA和ΦX174复制形式。这些切口有3′-OH和5′-P末端,而且是复制的活性点。用微球菌核酸酶引入的切口,产生的3′-OH和5′-P末端不是复制的活性点;它们抑制复制。无论切口的种类如何,形成复合物的聚合酶分子在数量上恰好等于切口数(图2中的插图)。图2中的沉降曲线,峰Ⅰ表示聚合酶分子结合到质粒的完整的双圆环形式,峰Ⅱ表示聚合酶分子结合到质粒的切口形式。
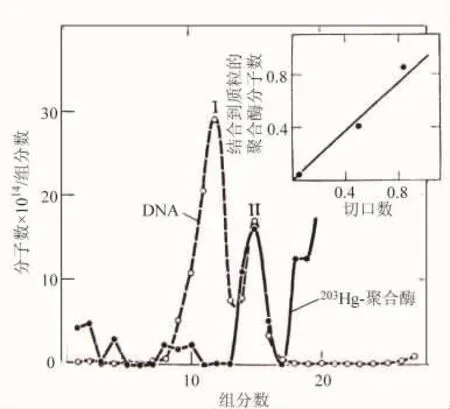
图2 切口质粒DNA结合到酶
在表1中总计了DNA结构与聚合酶的结合。在螺旋区域完全没有结合。沿着单股链到切口和末端有结合。在噬菌体T7的线性双链体DNA的情形下,我们的制剂平均每2个分子包含1个切口;这就解释了为何每个T7DNA分子结合2.6个酶分子。
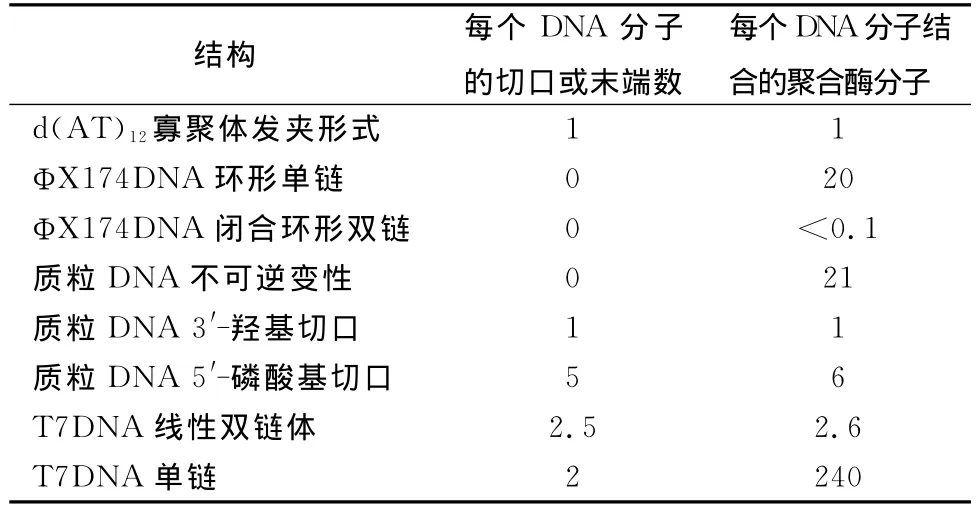
表1 DNA结构对DNA结合到DNA聚合酶的影响
1.2 酶的多重功能
为了在酶的活性中心开始构建一个可能发生的许多操作的模型,在这里他们想把结合实验的结果与他们已经研究的酶的催化性质联系起来。这些操作包括:①DNA链按照核苷酸的聚合作用沿5′→3′方向生长;②DNA链从3′-OH端水解(3′→5′方向);③DNA链从5′端水解(5′→3′方向);④DNA链从3′端进行焦磷酸解作用;⑤无机焦磷酸(PPi)与脱氧核糖核苷三磷酸的末端焦磷酸基团交换。
他们把酶活性中心画为特别适宜于识别并调节几个核苷酸结构的多肽面(图3)。他们提出在这个活性中心内至少有5个主要的部位:① 有一个部分模板链的部位(template site)。这个区域结合形成碱基对的这段链,它的任一边有几个核苷酸长度,这条链以特定的极性取向,很可能是结合环形单链DNA的部位。但是我们不能确定,这个部位是认可伸展的构型还是认可紧密、重叠的螺旋构型。②有一个生长链的引物部位(primer site),这个引物的取向与模板的极性取向相反。③有一个特别认可的具有核苷酸的3′-OH基团的引物末端部位(primer terminate site)。以后我们将讨论这个区域,作为3′-OH-末端引物链(3′→5′方向)水解的和焦磷酸解的剪切部位。④有一个三磷酸的部位(triphosphate site)。⑤有一个以后要考虑的,为5′-P-端链(5′→3′方向)的水解剪切提供的附加的部位。
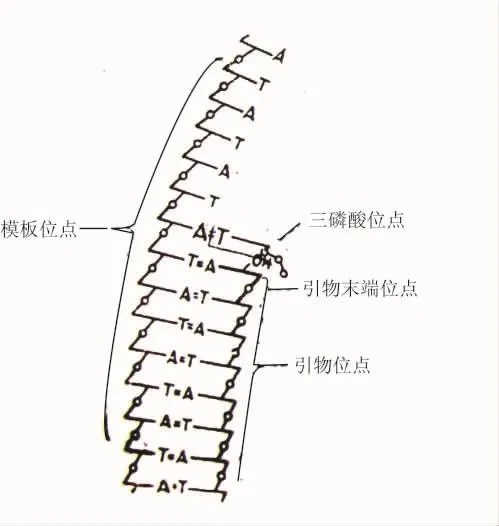
图3 在DNA聚合酶的活性中心内的部位
1.3 聚合阶梯
当一个线性双链体从3′-OH末端部分地被降解时,就确定的外切核酸酶作用的例子而言,这些被剥去的部分被所有的DNA聚合酶以极大的可能性修复,这是如何完成的?
在接近引物末端核苷酸上的3′-OH基团处结合三磷酸并调整其方向,以便三磷酸与引物能够进行直接的连接并与模板形成一个碱基对(图4)。当形成正确的碱基对时,发生了由引物端的3′-OH对三磷酸最内部的磷进行亲核的攻击。由于下述的理由,一个似乎合理的模型假设引物链相对于酶的运动或平移是与二酯键的形成一致的。当这个引物端平移进二酯键中,失去它的3′-OH基团时,它就不再占有这个引物端位置。通过整条链的运动,旧的引物端被一个具有末端3′-OH基团且保持在引物端部位的新加入的核苷酸取代。(现在这个新的引物端准备攻击另一个三磷酸,并加上下一个核苷酸)。只有当磷酸二酯键的形成完成时无机焦磷酸才被置换,而链的运动正好把新加的核苷酸转移到引物端部位。
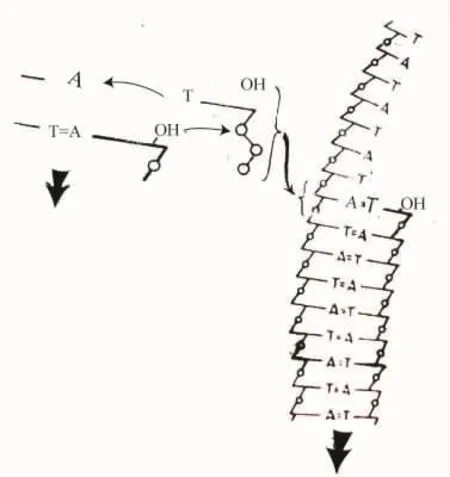
图4 聚合阶梯的机制
2 聚合酶亚单位结构的证实
2.1 聚合酶分子量的测定
1969年6月,科恩伯格在斯坦福大学医学院生物化学系的同事 T.M.Jovin,P.T.Englund和L.L.Bertsch在The Journal of Biological Chemistry第244卷上发表“DNA的酶合成:(26)均匀DNA聚合酶的物理和化学研究”的论文[2]。在这篇文章中,他们通过沉降平衡确定了DNA聚合酶的相对分子量是109 kDa。
沉降平衡法是从密度梯度离心技术发展起来的一种方法。样品放入离心管内的溶液中,进行离心,溶液沉降,几小时后达到平衡,则形成稳定的密度梯度。样品在离心管中运动,样品中密度大于周围溶液密度的向管底沉降,而密度小于周围溶液密度的样品向液面浮起。这两种运动一直延续到所有样品都到达某一平衡位置,形成平衡区带。
科恩伯格的同事们利用沉降平衡法计算DNA聚合酶分子量的公式如下:
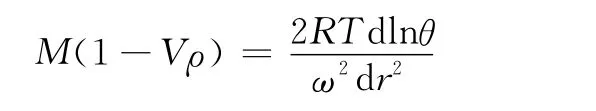
上式中M是分子量,V和ρ分别是比容和溶液密度,T是绝对温度,R是气体常数,ω是角速度,r是离转轴的径向距离,θ或△Z′是从零干涉水平上垂直条纹的位移,或者是△ji/2,在半条纹单位内从零浓度的位移。为了求得微商,他们按照实验值绘出了lnθ对r2的关系曲线(图5)。图5A中所显示的是在T=279.2 K,ω=20410rpm的稀释缓冲液中获得的数据,显示出没有偏离直线性。图5B所显示的是在T=298 K,ω=27690rpm时,在包含6M胍盐酸和0.3M的二硫基乙醇溶液中获得的的数据,同样没有偏离直线性。在细胞内达到的蛋白质的最大浓度是8 mg/L。在两种情形下,获得的分子量都是109 kDa。
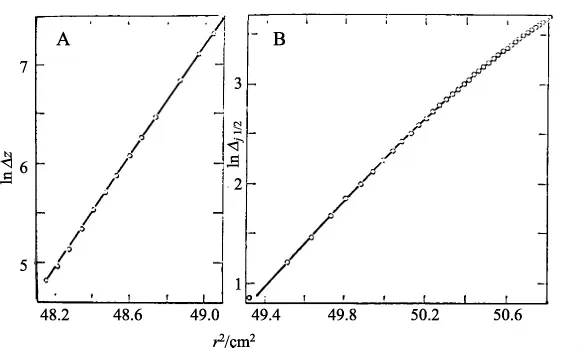
图5 DNA聚合酶的平衡沉降
2.2 聚合酶的活性片段
1969年,科恩伯格和他在斯坦福大学医学院生物化学系的同事D.Brutlag,M.R.Atkinson和P.Setlow在Biochemical and Biophysical Research第37卷上发表题为“由蛋白水解剪切产生的DNA聚合酶的活性片段”的论文。文章一开始就说:“大肠杆菌DNA聚合酶是具有109 kDa分子量的单条多肽链,在单个活性中心催化几种反应。在DNA引物的3′端有与聚合反应密切联系的3′→5′外切核酸酶活性,焦磷酸交换和焦磷酸解作用。脱氧核苷三磷酸底物的结合部位被认为是靠近有缺口的DNA的催化5′→3′外切核酸酶降解的另一个酶的位置。本文叙述了通过蛋白水解作用把DNA聚合酶剪切成两个分子量为76 kDa和34 kDa的片段,这个较大的片段保持了聚合酶活性和3′→5′核酸酶活性,但是没有5′→3′核酸酶功能。”[3]
他们使用聚丙烯酰胺凝胶电泳分离聚合酶并测定其分子量。电泳是指带电粒子在直流电场中移动的现象。不同的成分在均一的缓冲液中分离成独立的区带,可以用染色的方法显示出来,如果用光密度计扫描可得出一个个互相分离的峰。聚丙烯酰胺凝胶是一种多孔凝胶,它根据蛋白质的两个参数来进行分离:①蛋白质的净电荷;②蛋白质的分子大小。聚丙烯酰胺俗称“电泳分子筛”,具有很高的分辨率[4]。
如果在上述凝胶电泳系统中加入阴离子去污剂SDS(十二烷基硫酸钠),那么蛋白质就能解离成亚基。它们的电泳行为只与其分子大小有关,而与它所带的电荷无关,故可用这一性质测定蛋白质亚基的分子量。经验表明:在一定的凝胶浓度下,其泳动率与分子量的对数呈直线关系。因此先由一些已知分子量的蛋白质作出标准曲线,通过对比,便可测知未知蛋白质样品的分子量,还可判断出是否存在亚基[4]。
他们指出:“大肠杆菌DNA聚合酶是具有109 kDa分子量的单条多肽链的证据包括SDS-聚丙烯酰胺凝胶电泳分离(图6A)……从液氮储存器取出酶的样品并在0℃时保持在缓冲液中达四周就会产生两条带(图6C)。主要的条带处在分子量为76 kDa的位置,连同处在分子量为34 kDa的微弱带。这个分离的酶仍然显示出具有初始测定的全部聚合酶活性。在用分离的酶合成多聚d(A-T)中聚合作用有助于完成反应,而且不降解d(A-T)产物,像使用正常的酶一样。这就表明在分离的聚合酶中不存在核酸酶功能。”[3]
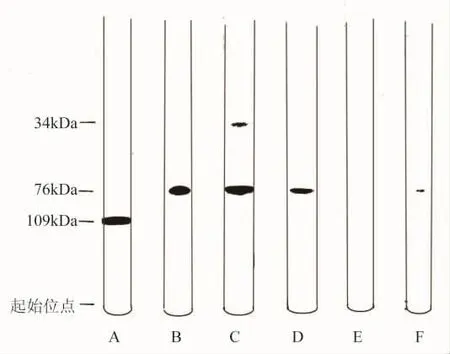
图6 DNA聚合酶的SDS-聚丙烯酰胺凝胶电泳图
聚合酶的分离是由蛋白酶催化肽键水解的结果。他们研究了几种包括枯草杆菌提取物、胰蛋白酶、胰凝乳蛋白酶和枯草杆菌蛋白酶BPN的蛋白酶对聚合酶的作用(图6C,D,E和F)。就每种情形下出现的聚合酶的剪切而论,在分离的聚合酶中看到的降解的图样最像用枯草杆菌提取物处理的情形。超过90%的聚合酶被剪切,产生76 kDa和34 kDa分子量的片段与少许中间大小的产物(大约53 kDa)。胰蛋白酶也产生76 kDa分子量片段作为主要的产物,除此之外还有大量53 kDa分子量的物质。胰凝乳蛋白酶和枯草杆菌蛋白酶BPN对限定剪切的产生影响很小,虽然在降解的产物中能够辨认出76 kDa分子量的片段。
他们通过凝胶过滤分离了由枯草杆菌提取物,蛋白水解聚合酶的产物。被分离的酶显示出聚合酶活性的增大和在5′→3′方向上核酸酶活性的减少。表2中聚合酶活性的速率表示为每个酶分子每分钟结合的核苷酸分子数。核酸酶活性的速率表示为每个酶分子每分钟释出的核苷酸分子数。用胰蛋白酶处理也引起聚合酶活性的增加(约增加了1.5倍)。通过长时间的处理,它自身分子量降解为76 kDa,聚合酶活性降低。
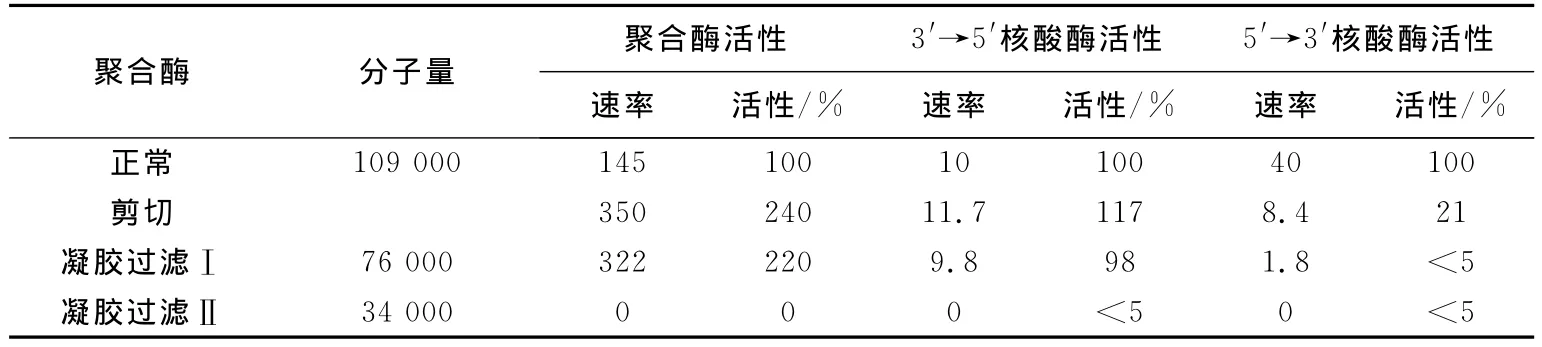
表2 分离前后聚合酶活性的比较
蛋白水解聚合酶的凝胶过滤是这样进行的:先用DNA聚合酶处理枯草杆菌提取物,而后经G-75交联葡萄糖柱层析这些产物,再用磷酸钾缓冲液洗脱。洗脱的结果如图7所示,图中在每个峰下合并的组分用Ⅰ和Ⅱ表示。组分Ⅰ的电泳显示出它是具有76 kDa分子量的单条带(﹥90%)。聚合酶功能和3′→5′外切核酸酶功能属于分子量为76 kDa的片段(表2)。当时在任何一个分离组分中没有探测到5′→3′核酸酶活性。后来的实验证明分子量为34 kDa的小片段没有聚合酶活性只包含5′→3′外切核酸酶活性。

图7 蛋白水解聚合酶的凝胶过滤
3 聚合酶结构的确立
1985年2月耶鲁大学分子生物物理和生物化学系教授T.A.Steitz等人在Nature上发表了题为“大肠杆菌DNA聚合酶Ⅰ的大片段与胸苷一磷酸复合的结构”的论文。宣布他们获得了第一个聚合酶的晶体结构,大肠杆菌DNA聚合酶Ⅰ(polⅠ)的蛋白水解的大片段。指出:“这个分子是相对分子量为103000(即103 kDa)的单体,它有三种酶的活性:DNA聚合酶,被认为是消除错配核苷酸的3′→5′外切核酸酶和5′→3′外切核酸酶。限定的蛋白水解把分子分解成两个片段;大的C端片段(Klenow片段)具有DNA聚合酶和3′→5′外切核酸酶活性,而小的 N端片段具有5′→3′外切核酸酶活性。”[5]
作者接着指出:“DNA聚合酶的活性位置和校正错配碱基的3′→5′外切核酸酶的活性位置在某种程度上在PolⅠ上是分离的。聚合酶活性位置结合着包含单链5′延伸的双链DNA和脱氧核苷三磷酸(dNTP)。虽然脱氧单磷酸(dNMP)也结合到polⅠ,但是它们并不冲突,因为它们的结合位置是分开的。由产物的抑制性推测起来,因为dNMP抑制3′→5′外切核酸酶活性而不抑制聚合酶活性,dNMP好象是结合到的3′→5′外切核酸酶活性位置。”[5]
1987年8月Steitz等人在Trends in Biochemical Sciences上发表题为“DNA聚合酶Ⅰ:从晶体结构到功能的遗传学研究”的论文。他们进一步指出:“结构的、生物化学的和遗传学研究的结合导致polⅠ有三个领域,每个领域担负着一种分离的酶的活性的结论(图8)。polⅠ的限定的蛋白水解除去了包含5′→3′外切核酸酶活性的34 kDa的N端区域。留下的68 kDa大片段(Klenow片段)具有我们最感兴趣的聚合作用和删除活性。如下所述,Klenow片段结构有对应于性质不同的聚合作用和删除功能。这两个活性位置大约相距30Å,引起了两种活性以什么方式一起工作的一些有趣的问题。”[6]
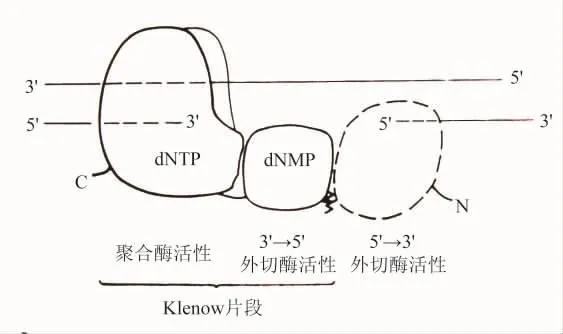
图8 大肠杆菌DNA聚合酶Ⅰ的近似的区域结构示意图(实线表示实验上确定的Klenow片段结构;虚线表示由polⅠ的蛋白水解剪切产生的小片段可能的定位)
3.1 Klenow片段的结构
在上述1985年的论文Klenow片段的结构一节中,他们把Klenow的晶体结构分成大区域和小区域两个领域。明确指出大区域最明显的结构特征是在标记I和O的螺旋之间存在一条很大的裂缝,类似于右手的形状,手指和拇指对应于O螺旋和I螺旋(图9)。
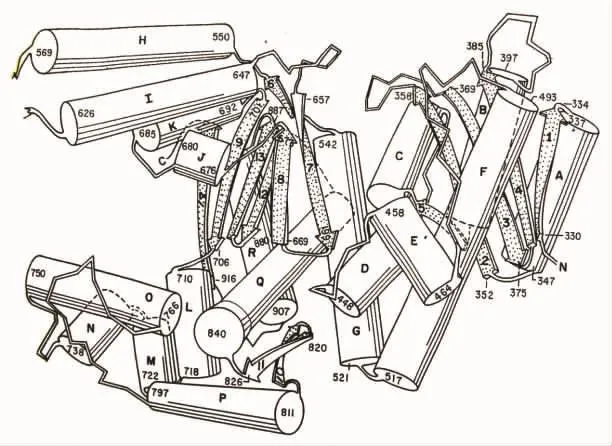
图9 Klenow片段的三维结构(圆柱体表示α螺旋;箭头表示β折叠片构型;I和O螺旋间的裂缝很可能是与DNA相互作用的位点)
他们写道:“这个大区域的400个氨基酸(521→928残基)主要是以α螺旋构型出现,形成包含一条很深裂缝的结构,类似于握着一根杆的右手的形状(图9)。6条绞合的反平行的β片形成了裂缝的底部。在任何一边α螺旋的大的伸出部分形成了它的边。这条裂缝20~24Å宽,25~35Å深,具有伸进裂缝的两个α螺旋,J和K。这个裂缝的一条边是比较长的(是50Å而不是20Å),能够与右手弯曲的手指比较。主要由两条长的α螺旋I和H形成了其他的壁,I和H从蛋白质伸出并像一个拇指附着于裂缝。”[5]
接着指出小区域具有结合脱氧核苷单磷酸(dNMP)和二价金属离子的功能。他们说:“我们发现这个片段折叠成两个截然不同的区域(图9)。较小的片段大约是polⅠ序列中的头200个残基(324→517)。形成在两边上具有α螺旋,中心是多数平行的β折叠片。脱氧核苷单磷酸结合到靠近β链的羧基端区域。这个小区域包含了一个与蛋白质和脱氧胸苷单磷酸(dTMP)相互作用的结合金属离子。把掺入 Zn2+,Mn2+,Sm3+或 Mg2+的晶体和没有金属离子的晶体比较,揭示了能够结合这些金属离子中任何一种离子的牢固的结合位置……被Asp355(天冬氨酸355),Glu357(谷氨酸357)和Asp501的羰基(carboxylate group)把金属离子结合到蛋白质上。在dTMP复合物中,这个5′磷酸基给金属离子提供了第4个配体。”[5]
3.2 DNA的结合部位
关于DNA结合部位的研究来自于对各种与引物-模板接头结合的DNA聚合酶的原子结构的研究。这些结构揭示了DNA底物位于一个大的、形似一只半握的右手的裂缝之中。在1985年的论文“DNA结合部位”一节中,Steitz T A等人明确指出DNA结合部位位于I和Q之间的大裂缝之中。他们写道:“这个大裂缝的尺寸和形状表明它结合DNA合成的双链体产物。如果J和Kα螺旋部分地处于较大的裂缝中(图10),B-DNA就适合于这个裂缝。虽然在J和K螺旋之间又有结构上和功能上的类似,而且这两个螺旋的特色为一些调节蛋白所共有,但是似乎没有稳固的结构上的相应物。当聚合酶沿着DNA滑动时,要求蛋白质做螺旋运动,在PolⅠ中两个螺旋可能对于把DNA的平移位置附于蛋白质上起作用。这个裂缝的一边大约是50长,可能对DNA螺旋轴的定向起作用。它也能够结合单链模板。”[5]
他们指出:“polⅠ的结构表明它具有持续合成能力的机制。也就是说,在它断绝和DNA底物的关系以前,照其他聚合酶反应中共有的样子,它具有把许多核苷酸结合到DNA的能力。在我们的模型中柔性地附着在I和H螺旋的顶部50个残基子域的定位表明在DNA结合之后这个小的子域阻塞了这个裂缝。于是,如果这个蛋白质隔绝了它的DNA底物,DNA产物解离的速率可能很慢,如果比聚合速率慢很多,就可能导致连续的聚合。这个酶就可能简单地滑动到在聚合阶梯的新的3′端。”[5]
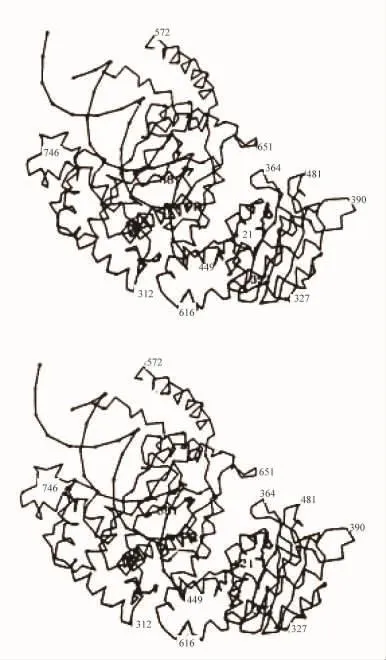
图10 双链DNA的结合位置(用连接磷位置的线表示双链DNA,引物端可能的定位用PR标记)
他们继续写道:“对于引物链3′端的定位虽然我们没有证据,在图10中提出的位置好象是可能的。这个引物端处在以下的情况:①尽可能靠近dTMP;②与蛋白质互相配合;③允许合成的单链模板链和蛋白质之间的相互作用;④处在最靠近N末端的DNA端,像切口平移所需要的从N端取出的小片段。作为一种选择,我们试图用附着到核苷单磷酸的观察位置的DNA3′端建立一个酶-DNA 模型。”[5]
3.3 3′→5′外切核酸酶活性位置
在1987年的论文[6]的“3′→5′外切核酸酶活性位置”一节中,指出:“这个晶体结构显示dNMP结合到Klenow片段的小区域(图11)。因为脱氧核苷单磷酸抑制3′→5′外切核酸酶反应(根据抑制产物推测),很有可能在晶体中dNMP的结合位置与在外切核酸酶活性位置中DNA3′端的位置一致。因此,这个dNMP磷酸基将表示在外切核酸酶反应中被切开的磷酸二酯键的位置。”[6]
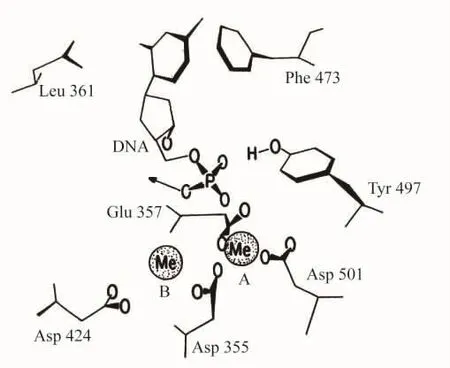
图11 具有结合分子dCMP的3′→5′外切核酸酶活性位置
Klenow片段结构的精制证实其它晶体学的数据,表明dNMP结合区域包含两个二价金属离子的位置(图11)。由Asp355,Glu357和Asp501的羰基与提供了第4个配体的dNMP 5′磷酸基协调一个金属离子(位置A)。Mg2+或者Zn2+甚至在dNMP不存在时也能够结合到晶体中的这个位置。第2个二价金属离子定位在dNMP磷酸基和Asp424的羰基之间(位置B)。用EDTA(乙二胺四乙酸)除去结合到晶体的dNMP从而分离二价金属离子是与早期的平衡透析研究一致的。
为了研究两个金属离子的作用,他们通过定位诱变产生两个突变蛋白质。由于金属B和Asp424相联的边链邻近dNMP磷酸基,他们引起突变Asp→Ala424。当纯化到均匀时,这个最后得到的蛋白质具有野生型聚合酶活性的水平,但是基本上没有外切核酸酶活性。对与dNMP复合的这个蛋白质的高分辨晶体学分析表明:dNMP和金属A的结合是正常的。与野生型复合物唯一的差别是Asp424羰化物和在位置B处金属离子的不存在。于是在Asp→Ala424时,蛋白质中外切核酸酶活性的丢失好象是除去金属位置B的直接的结果,从而提出在催化中这个金属离子起着直接的作用。他们还通过造成双突变Asp→Ala355,Glu→Ala357,消除了金属A位置的两个配体。这个突变的蛋白质也具有聚合酶活性,但是没有外切核酸酶活性。低分辨晶体学的研究表明,大的和小的区域结构都未被干扰;然而这个晶体没有结合dNMP,或者没有结合在位置A和B中任何一个金属离子。因为在金属B不存在时能够结合dNMP(像由Asp→Ala424突变所示的一样),这个结果意味着在底物结合中包含金属位置A,虽然在催化中没有把附加的作用排除在外。
在1987年论文的的最后一节中,作者写道:“通过在聚合酶反应中选择正确的dNTP和通过删除错误结合的碱基获得了由polⅠ合成的精确的DNA。科恩伯格小组证实了3′→5′外切核酸酶反应的删除能力,他们说明这个活性能够排除在引物端错配的碱基对。在聚合酶反应中dNTP选择的机制和其他删除步骤可能的存在现在还不清楚。因此我们将把我们的讨论限制在3′→5′外切核酸酶反应的删除作用上。”[6]
他们接着指出:“现在在研究中的Klenow片段-DNA共晶体显示出在外切核酸酶活性位置中的DNA 3′端并允许我们推测这个‘删除复合物’的结构。在共晶体中看到的单链DNA端能够很容易地联想到已经建立的在大区域上结合裂缝的引物链模型(图12)。”[6]
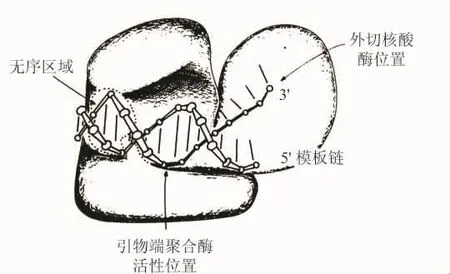
图12 当引物链的3端处在外切核酸酶位置(exonuclease active site)时Klenow片段结合到DNA分子上的示意图(引物端从聚合酶活性位置到外切核酸酶活性位置运动的范围用加黑的键表示)
3.4 聚合酶结合DNA结构的确定
1993年,Steitz等人在Science第260卷上发表题为“DNA聚合酶ⅠKlenow片段结合双链体DNA”的论文。[7]在引言中作者首先概述了与双链体DNA共结晶的大肠杆菌DNA聚合酶Ⅰ的Klenow片段。他们说:“我们把11个DNA碱基对置入处在与裂缝成直角的沟内,这条裂缝包含了聚合酶活性位置并与3′→5′外切核酸酶区域相邻接。当Klenow片段与DNA结合时,一个以前被认为是无序的区域成为更加有序的区域,与两条螺旋一起朝着3′→5′外切核酸酶区域运动形成这条结合沟。在单链区域的3′端,以三个核苷酸长度结合在外切核酸酶活性位置。虽然这个共结晶的结构好像是一个编排的复合物,但是它表明这条引物链从3′→5′外切核酸酶区域的方向接近聚合酶的催化位置,而这条双链DNA产物可能弯折到进入包含聚合酶催化位置的裂缝。[7]”接着明确指出:“在这个晶体结构中,催化这些反应的两个活性位置大约相距35Å。”又说:“聚合酶反应的催化位置定位在这个大裂缝的底部。这个区域包含了在聚合酶序列中许多高度保守的残基。”[7]
最后,他们在观察复合物晶体结构的基础上,描绘了一个既删除又聚合复合物模型,如图13所示。他们提出:“这个引物-模板(primer-template)结合在聚合酶裂缝中。在聚合酶裂缝中,这条引物链碱基是与模板的碱基配对的,而它的3′端是靠近结合到催化重要的Asp882,Glu883和Asp705的两个二价金属离子。由于观察到的裂缝狭窄需要双链体引物端的某些变形或蛋白质的运动。引物链的位移必然把模板链带到引物端,使螺旋区域缩短,形成与拇指(thumb)子域对应的裂缝的壁。虽然这个DNA位于聚合酶裂缝内,像在原来的模型中一样,但是这个假设的DNA合成的方向是相反的。包含了引物端的这个双链体DNA位于邻近外切核酸酶区域的裂缝边缘,而这条单链模板链从这条裂缝的的顶部进入。”[7]
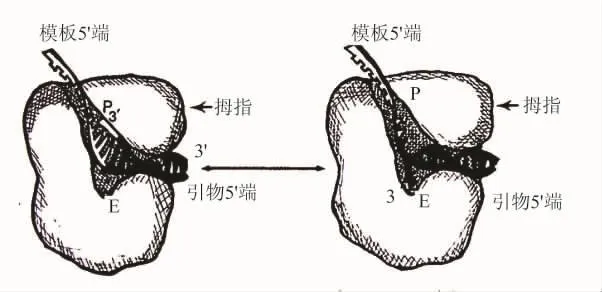
图13 DNA的结合位置模型(聚合酶活性位置P;3′到5′外切核酸酶活性位置E)
关于聚合酶裂缝的弯折他们作了如下的说明:“相对于聚合酶裂缝1在裂缝2中DNA螺旋轴的角度是料想不到的。如果人们构造一个模型,要保持多数观察到的在双链体DNA和蛋白质之间的缩短,那么构筑的这个DNA模型大约需要弯折80°才能进入聚合酶裂缝(图13)。双链体DNA的这种诱导蛋白的弯折是知道的。这个分解代谢物基因激活蛋白与DNA复合物的晶体结构表明,主要通过两个43°弯折达到 DNA弯折90°。”[7]
3.5 聚合酶复合物结构的确定
1998年,哈佛大学医学院生物化学和分子药物系教授Doublie等人在Nature上发表题为“噬菌体T7DNA复制的晶体结构——以2.2最短距离分辨的复合物”的论文。介绍了在聚合酶活性位置处从与引物-模板和核苷三磷酸复合的噬菌体T7中提出的最短分辨距离为2.2的复制DNA聚合酶的晶体结构。
在本文“复合物结构的确定”一节中,作者写道:“T7DNA有一个在polⅠ家族中为其他聚合酶特有的结构,具有氨基(-NH2)端的3′→5′校对外切核酸酶区域和羧基(COOH)端的聚合酶区域。这个聚合酶区域有一个使人联想起右手的形状,在右手中这个手掌、手指和拇指形成导致聚合酶活性位置的DNA-结合沟(图14)。这个引物模板的定位,使3′引物端处在手指和拇指的汇合处,靠近ddGTP。这个手掌形成了聚合酶活性位置的基底,显示出三个严格保守的和功能上重要的酸性残基。两个金属离子通过两个保守的羰化物和依次连接这个结合ddGTP的磷酸基而保持在这个位置(图15)。这些手指围在DNA结合沟端部的外边,引起与结合核苷酸的许多相互作用……这个结构的保守性在手掌中是最明显的,在那里T7DNA聚合酶的β-链9,12和13很好地叠在类似的Klenow片段的链上。这个3′→5′外切核酸酶区域处在远离手指的DNA结合沟的末端,类似于在Klenow片段中的定位。在T7DNA聚合酶中,嵌入大拇指顶部71个残基形成一个结合到硫氧还蛋白扩展的环上,悬在退出聚合酶的双链体DNA上。”[8]
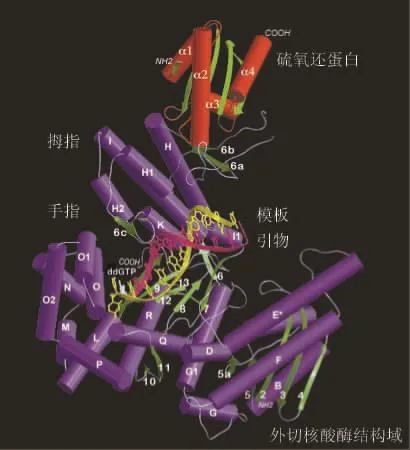
图14 T7DNA聚合酶复合物的结构
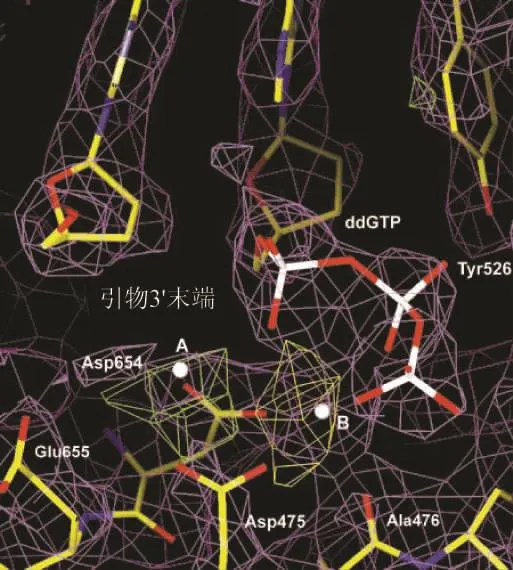
图15 结合核苷酸的两个金属离子的连接作用
2008年,在沃森(J.D.Watson)等人编著的《基因的分子生物学》一书中,根据Doublie等人的实验结果描绘出DNA与聚合酶结合的右手模型图。他们写道:“关于DNA聚合酶如何催化DNA合成的分子解释,来自于对各种引物-模板接头结合的DNA聚合酶的原子结构的研究。这些结构揭示了DNA底物位于一个大的、形似一只半握的右手的裂缝之中(图16),依据对手的类比,聚合酶的3个结构域被称为拇指、手指和手掌。”[9]
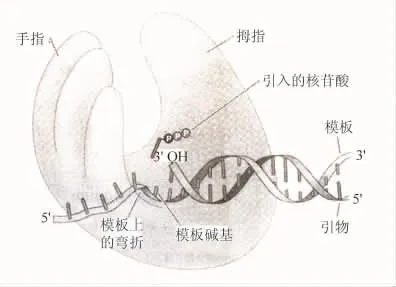
图16 模板DNA穿过DNA聚合酶的路径示意图
关于聚合酶这三个结构域的作用,他们指出手掌域由β折叠片构成,含有催化位点的基本元件,这一区域的2个二价金属离子可以改变dNTP和3′引物端周围的化学环境,起催化作用。除此之外,还负责检查新加入的核苷酸碱基配对的准确性。手指域对催化也很重要。一旦引入的dNTP与模板之间形成正确的碱基配对,手指域即发生移动,包围住dNTP。聚合酶手的这种闭合形式可以使引入的核苷酸与催化性的金属离子密切接触,从而促进催化反应。手指域还与模板结合,使模板的磷酸二酯键骨架在活性部位后立即产生约90°的回折,此弯曲仅使催化位点上引物后的第一个模板碱基暴露。模板的这种构象避免了在下一个核苷酸添加时可能会造成的对配对的模板碱基选择的混淆。拇指域与催化的关联不紧密,而是与最新合成的DNA相互作用,维持引物与活性部位的正确位置,以及聚合酶与其底物的紧密连接。这种连接有助于聚合酶每次与引物-模板接头结合时,具有添加许多dNTP的能力。
(2011年3月17日收到)
[1]KORNBERG A .Active center of DNA polymerase[J].Science,1969,163:410-1418.
[2]JOVIN T M,ENGLUND P T,BERTSCH L L.Enzymatic synthesis of deoxyribonucleic acid:XXVI.physical and chemical studies of a homogeneous deoxyribonucleic acid polymerase[J].The Journal of Biological Chemistry,1969,244(11):2996-3008.
[3]BRUTLAG D,ATKINSON M R,SETLOW P,KORNBERG A.An active fragment of DNA polymerase produced by proteolytic cleavage [J].Biochemical and Biophysical Research,1969,37:982-989.
[4]何忠效.生物化学实验技术 [M].北京:化学工业出版社,2004:168,175,176,356,357.
[5]OLLIS D L,BRICK P,HUONG R,STEITZ T A.Structure of large fragment of Escherichia coli DNA polymerase Ⅰ complexed with dTMP[J].Nature,1985,313:762-766.
[6]JOYCE C M,STEITZ T A.DNA polymeraseⅠ:from crystal structure to function via genetics[J].Trends Biochem Sci,1987,12:288-292.
[7]BEESE LORENA S,DERBYSHIRE V,STEITZ T A.Structure of DNA polymeraseⅠKlenow fragment bound to duplex DNA [J].Science,1993,260:352-355.
[8]DOUBLIE S,TABOR S,LONG A M,et al.Crystal structure of a bacteriophage T7 DNA replication complex at 2.2Åresolution[J].Nature,1998,391:251-258.
[9]沃森J D,等,编著.基因的分子生物学(第六版)[M].杨焕明,等,译.北京:科学出版社,2009:203-207.
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
肝博士(2022年3期)2022-06-30
中等数学(2022年1期)2022-06-05
无机化学学报(2020年7期)2020-07-20
中国预防兽医学报(2020年2期)2020-06-01
三农资讯半月报(2020年8期)2020-05-13
中国饲料(2019年19期)2019-03-25
中等数学(2018年4期)2018-08-01
中学数学研究(广东)(2018年23期)2018-03-05
