
反相高效液相色谱法测定奥沙普嗪分散片中奥沙普嗪含量
2011-01-24 06:48吴飞跃
中国药业 2011年10期
吴飞跃,刘 健
(1.浙江省舟山医院,浙江 舟山 316000; 2.浙江大学医学院附属第一医院,浙江 杭州 310003)
反相高效液相色谱法测定奥沙普嗪分散片中奥沙普嗪含量
吴飞跃1,刘 健2
(1.浙江省舟山医院,浙江 舟山 316000; 2.浙江大学医学院附属第一医院,浙江 杭州 310003)
目的建立测定奥沙普嗪分散片中奥沙普嗪含量的高效液相色谱法。方法采用反相高效液相色谱法,以Hypersil C18柱(200 mm×4.6 mm,5μm)为色谱柱,以0.01mol/L磷酸二氢钾溶液(磷酸调pH至2.5)-乙腈 (40∶60)为流动相,检测波长为286nm,流速为1.0mL/min,柱温30℃。结果奥沙普嗪质量浓度在4.0~24.0 μg/mL范围内与峰面积线性关系良好,高中低3种质量浓度的回收率为97.6% ~101.3%,RSD为1.1% ~1.8%。结论所建立的方法快速、准确、简便,适用于奥沙普嗪分散片的质量控制。
奥沙普嗪;高效液相色谱法;含量测定
1 仪器与试药
高效液相色谱仪(日本岛津),包括LC-10ATvp泵,SPD-10Avp型紫外检测器;BP 221S电子分析天平(德国赛多利斯);KQ-250B型超声波清洗器(昆山市超声仪器有限公司)。奥沙普嗪化学对照品(含量为99.9%,天津金世制药有限公司);奥沙普嗪分散片(沈阳富东制药有限公司,规格为0.2 g/片);乙腈为色谱纯(美国Fisher公司),水为重蒸馏水,其余试剂均为分析纯。
2 方法与结果
2.1 色谱条件与系统适用性试验
色谱柱:Hypersil C18柱(200 mm ×4.6 mm,5 μm,大连依利特);流动相:0.01 mol/L磷酸二氢钾溶液(磷酸调pH至2.5)-乙腈(40 ∶60);检测波长:286 nm;流速:1.0 mL/min;柱温:30 ℃;进样量:20 μL。理论板数按奥沙普嗪峰计算应不低于2 000。
2.2 测定方法
取本品10片,精密称定,研细,精密称取适量(约相当于奥沙普嗪60 mg),置100 mL量瓶中,加流动相适量,超声处理30 min,放冷,加流动相定容,摇匀,滤过,取续滤液1 mL,置50 mL量瓶中,加流动相稀释至刻度,作为供试品溶液,精密量取20 μL注入液相色谱仪,记录色谱图。另取奥沙普嗪对照品适量,精密称定,加流动相溶解,制成每1 mL含奥沙普嗪12 μg的溶液,作为对照品溶液,同法测定。按外标法以峰面积计算,即得。
2.3 方法学考察
专属性考察:照2.2项下方法操作,供试品溶液、对照品溶液和空白辅料溶液的色谱图见图1。可见,流动相、辅料在奥沙普嗪峰处无干扰,奥沙普嗪峰与相邻杂质峰的分离度符合要求。
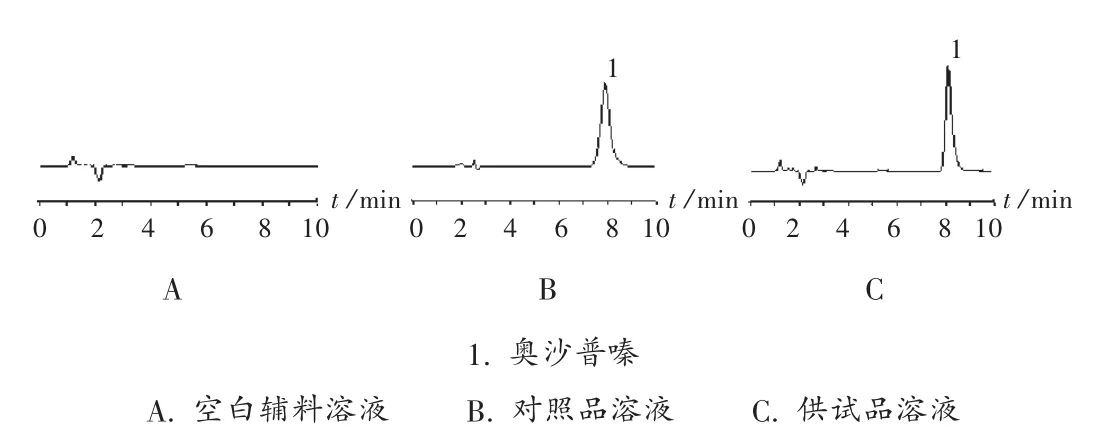
图1 高效液相色谱图
线性关系考察:取奥沙普嗪对照品适量,精密称定,用流动相制成质量浓度为 4.0,8.0,12.0,16.0,24.0 μg/mL 的系列溶液,照2.2项下方法测定,记录色谱图。以峰面积(Y)对质量浓度(X)进行线性回归,回归方程为 Y=25 837 X -426.1,r=0.999 9(n=5)。结果表明,奥沙普嗪质量浓度在4.0~24.0 μg/mL范围内与峰面积线性关系良好。
精密度试验:取奥沙普嗪对照品适量,精密称定,用流动相制成含奥沙普嗪 8.0,12.0,16.0 μg/mL 的溶液,分别精密吸取 20 μL 注入液相色谱仪,重复进样5次。结果低、中、高质量浓度溶液峰面积的 RSD 分别为 1.11% ,1.64% ,0.98%(n=5),表明仪器精密度(进样与峰面积检测精密度)良好。
重复性试验:取同一样品,照2.2项下方法制备供试品溶液6份,按外标法测定。结果奥沙普嗪的平均标示百分含量为99.17%,RSD为0.31%(n=6),表明方法重现性良好。
稳定性试验:照2.2项下方法制备供试品溶液,在室温下于不同时间(1,2,3,4,6,8 h)分别进样 20 μL。结果峰面积的 RSD=1.98%(n=6),表明供试品溶液在室温下8 h内稳定性良好。
回收率试验:于空白辅料中,按处方量加入奥沙普嗪对照品适量,照2.2项下供试品溶液制备方法制备低、中、高3种质量浓度的溶液各3份,按外标法分别测定并计算回收率。结果见表1。
2.4 样品含量测定
取3批样品,照2.2项下方法测定。结果3批样品中奥沙普嗪的标示百分含量分别为 99.4% ,100.5%,100.1% 。
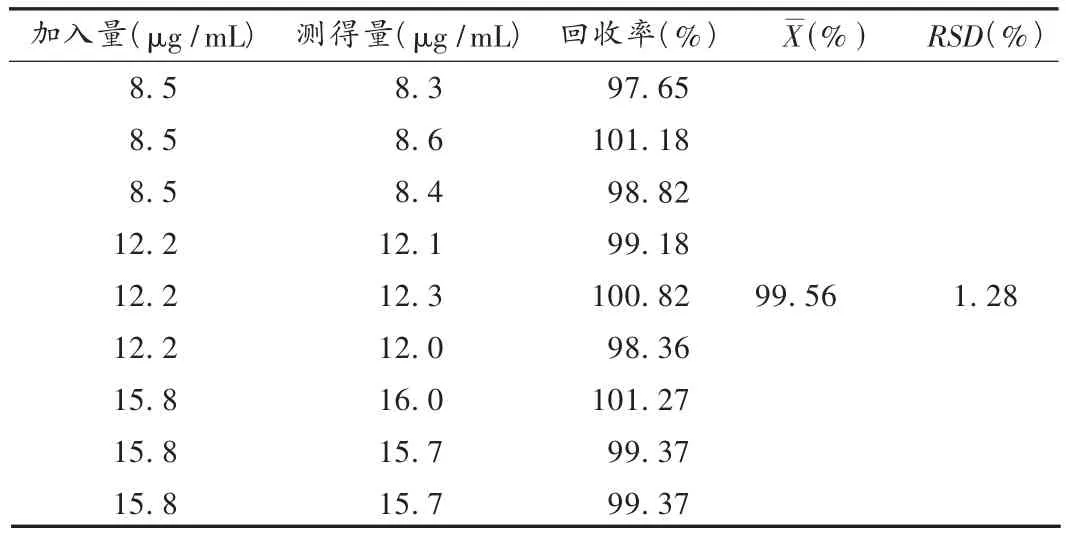
表1 回收率试验结果
3 讨论
目前2010年版《中国药典(二部)》收录了奥沙普嗪、奥沙普嗪肠溶胶囊,均采用紫外分光光度法测定奥沙普嗪的含量。高金波等[2]采用比色法测定了奥沙普嗪的含量。由于奥沙普嗪在合成中易带入一些有机杂质,可能对紫外法测定结果有影响。本试验采用了反相高效液相色谱法,比紫外分光光度法更准确、专属性更强。3批样品含奥沙普嗪均在标示量的99.4% ~100.5%范围内,供试品溶液主峰的保留时间与对照品溶液主峰的保留时间一致,而空白辅料溶液无此吸收峰,说明该法适合用于奥沙普嗪分散片的质量控制。
[1]李杨华.奥沙普嗪治疗类风湿性关节炎53例[J].医药导报,2002,21(11):727.
[2]高金波,李国清,张楠楠,等.用比色法测定奥沙普嗪的含量[J].黑龙江医药科学,2002,25(6):28 -29.
Determination of Oxaprozin Content in Oxaprozin Dispersible Tablets by RP-HPLC
Wu Feiyue1,Liu Jian2
(1.Zhoushan Hospital,Zhoushan,Zhejiang,China 316000; 2.The first affiliated hospital of Zhejiang Uviversity, Hangzhou,Zhejiang,China 310003)
ObjectiveTo establish a RP - HPLC method for the determination of Oxaprozin content in Oxaprozin Dispersible Tablets.Methods The RP -HPLC method was adopted on the Hypersil C18column(200 mm ×4.6 mm,5 μm)with UV detection wavelength at 286 nm,and the mobile phase consisted of acetonitrile-0.01 mol/L potassium dihydrogen phosphate solution adjusted to pH=2.5 with phosphoric acid(60 ∶40).The flow rate was 1.0 mL/min.The column temperature was 30 ℃.Results The calibration curve was linear within the range of 4.0 -24.0 μg/mL for oxaprozin,the average recovery rate was 97.6% -101.3% for three different concentrations of oxaprozin, RSD was 1.1% -1.8%.Conclusion This method is rapid,simple and accurate,which is suitable for the quality control of Oxaprozin Dispersible Tablets.
oxaprozin;HPLC;content determination
R927.2;R971+.1
A
1006-4931(2011)10-0037-02
2010-12-13)
猜你喜欢
当代水产(2022年4期)2022-06-05
口腔护理用品工业(2021年4期)2021-11-02
环境保护与循环经济(2021年7期)2021-11-02
食品安全导刊(2021年20期)2021-08-30
食品安全导刊(2021年20期)2021-08-30
中成药(2018年12期)2018-12-29
中成药(2017年10期)2017-11-16
中医研究(2014年11期)2014-03-11
中成药(2014年11期)2014-02-28
