
高效液相色谱法测定朱砂七中虎杖苷含量
2011-01-24 06:47杨海燕
中国药业 2011年10期
杨海燕,张 成
(1.陕西省药品检验所,陕西 西安 710061;2.陕西省医疗器械检测中心,陕西 西安 710061)
高效液相色谱法测定朱砂七中虎杖苷含量
杨海燕1,张 成2
(1.陕西省药品检验所,陕西 西安 710061;2.陕西省医疗器械检测中心,陕西 西安 710061)
目的建立测定朱砂七中虎杖苷含量的高效液相色谱法。方法采用Shimadzu VP-ODS色谱柱(250 mm×4.6 mm,5 μm),流动相为乙腈 -水(15∶85),流速 1.0 mL/min,柱温 30℃,检测波长 317 nm。结果虎杖苷峰与其他峰分离良好,进样量在 0.082 05~0.820 50 μg范围内与峰面积线性关系较好,平均加样回收率为98.70%,RSD=1.60%(n=6)。结论该法可以用于测定对朱砂七中虎杖苷含量,从而达到控制药材质量的目的。
朱砂七;高效液相色谱法;虎杖苷;含量
朱砂七为蓼科植物毛脉蓼 Polygonum Cillinerve(Nakai)Ohwl的干燥块根,临床上多用于胃炎、急性菌痢[1],疗效确切。现代药理学证实,本品具有抑菌、抗病毒及抗氧化活性作用[2-3]。近年来研究还发现,朱砂七含有较多的白黎芦醇、白黎芦醇苷(虎杖苷)等二苯乙烯类成分[4],因此其在抗肿瘤[5]、调节心血管和调血脂[6]方面的应用有待于进一步研究。为控制药材质量,笔者建立了测定朱砂七中虎杖苷含量的高效液相色谱图。
1 仪器与试药
LC-2010 HT型全自动高效液相色谱仪(日本岛津);AG ME 235S型电子分析天平,BS 224S型电子分析天平(德国赛多利斯);HH-4型数显恒温水浴锅(国华电器有限公司)。乙腈(色谱纯,美国Tedia公司),水(超纯水),其他试剂(分析纯);虎杖苷对照品(中国药品生物制品检定所,批号为111575-200301);朱砂七药材(部分为市售药材,部分由陕西盘龙药业有限公司提供)。
2 方法与结果
2.1 色谱条件与系统适用性试验
色谱柱:Shimadzu VP-ODS 柱(250 mm×4.6 mm,5 μm);柱温:30 ℃;流动相:乙腈 -水(15 ∶85);流速:1.0 mL/min;检测波长:317 nm;进样量:10 μL。在此色谱条件下,虎杖苷峰与其他峰分离良好,理论板数以虎杖苷峰计不低于3 000,色谱图见图1。
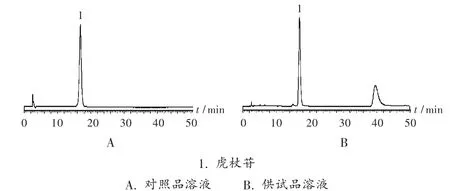
图1 高效液相色谱图
2.2 溶液制备
精密称取虎杖苷对照品10.94 mg,置100 mL量瓶中,用甲醇溶解并稀释至刻度,摇匀,再精密量取5 mL,置20 mL量瓶中,加甲醇至刻度,摇匀,作为对照品溶液。取朱砂七药材细粉0.2 g,置索氏提取器中,用乙酸乙酯回流提取至无色,提取液回收至干,残渣用甲醇溶解并定容至10 mL量瓶中,摇匀,用0.45 μm的微孔滤膜滤过,取续滤液,作为供试品溶液。
2.3 方法学考察
线性关系考察:分别精密吸取对照品溶液 3,5,10,15,25,30 μL,进样测定峰面积,以峰面积积分值为纵坐标、进样量(μg)为横坐标绘制标准曲线,回归方程为 Y=6.114 3×106X-4.904 2×105,r=0.993。结果表明虎杖苷进样量在 0.082 05 ~0.820 50 μg范围内与峰面积积分值有较好的线性关系。
精密度试验:精密吸取对照品溶液,重复进样6次,测定峰面积积分值。结果的 RSD为0.59%(n=6),表明仪器精密度良好。
稳定性试验:精密吸取同一供试品溶液10 μL,每隔2 h进样1次,测定峰面积积分值。结果的 RSD为1.3%(n=7),表明供试品溶液在12 h内基本稳定。
重复性试验:取同批朱砂七药材细粉6份,按供试品溶液制备方法制备溶液,测定含量。结果平均含量为1.099 8 mg/g,RSD为3.4%(n=6),表明方法重复性较好。
加样回收试验:取朱砂七细粉0.1 g,置索氏提取器中,分别精密量取0.101 6 mg/mL的虎杖苷乙酸乙酯溶液1,2,3 mL,按供试品溶液制备方法制备溶液并测定含量,计算回收率。结果见表1。

表1 虎权苷加样回收试验结果(n=6)
2.4 样品含量测定
测定了7批朱砂七药材的含量,结果虎权苷含量分别为2.82,2.66,1.10,3.65,1.54,6.90,1.81 mg/g,根据所测结果,规定每1 g药材含虎杖苷应不低于1.0 mg。
3 讨论
取加入虎杖苷对照品的流动相溶液,以流动相为空白,在200~400 nm波长范围内扫描,结果在317.00 nm波长处有最大吸收,故将检测波长定为317 nm。
在选择提取溶剂时,分别以乙醚、乙酸乙酯、甲醇回流提取依法测定。结果乙醚的提取率只有乙酸乙酯和甲醇提取率的1/10,乙酸乙酯和甲醇的提取率基本一致,但乙酸乙酯提取的样品杂质较少,虎杖苷与周围峰的分离明显好于甲醇提取,故选乙酸乙酯为提取溶剂。
选择提取方法时,以乙酸乙酯为溶剂,比较了超声(1 h)和索氏提取的效果,结果索氏提取的样品含量几乎是超声提取的4倍,故采用索氏提取法。
测定了 4 批市售饮片的含量,结果分别为 9.77,7.37,10.98,9.96 mg/g(含量均较高),炮制是否能使虎杖苷含量增加,有待进一步研究。由于所测药材和饮片的含量差异较大,当所测溶液含量过高(质量浓度超过0.082 mg/mL)时,将样品溶液稀释2倍或5倍体积后再测定,可以保证每一测量结果均在线性范围内。
[1]袁鲜云,武希桃.朱砂七抗炎及抗溃疡作用的实验研究[J].陕西中医学院学报,2003,26(2):48 -49.
[2]中医药管理局《中华本草》编委会.中华本草[M].上海:上海科学技术出版社,2000:650.
[3]Jong PL,Ryung SM,Ren BA,et al.Stilbenes from the roots of Pleuropterus ciliinervis and their antioxidant activities[J].Phytochemistry,2003,64(5):759.
[4]戚欢阳,张朝凤,张 勉,等.毛脉蓼化学成分及抑菌活性的研究[J].中国药学杂志,2005,40(11):819 -822.
[5]Jang M,Cai L,Geoge OU,et al.Cancer chemopreventive activity of resveratrol,a natural product devived from grapes[J].Science,1997,275:218 -220.
[6]舒仕瑜.白藜芦醇苷生物活性及药理作用[J].儿科药学杂志,2002,8(1):9-11.
Determination of Polydatin in Polygonum Cillinerve(Nakai)Ohwl by HPLC
Yang Haiyan1,Zhang Cheng2
(1.Shaanxi Provincial Institute for Drug Control, Xi'an, Shaanxi,China 710061;
2.Shaanxi Provincial Medical Equipment Examination Center, Xi'an, Shaanxi, China 710061)
ObjectiveTo establish the quantitative determination method ofpolydatin in Polygonum Cillinerve (Nakai) Ohwlby HPLC.Method Polydatin was determined by HPLC carried on the Shimadzu VP -ODS column(250 mm ×4.6 mm,5 μm)with the mobile phase of acetonitrile-water(15 ∶85)and the flow rate at 1.0 mL/min.The column temperature was 30 ℃ and the detection wavelength was 317 nm.Results The peak of polydatin was separated well with other peaks,showing the better linear relation between the sample size in the range of 0.082 05 - 0.820 50 μg with the peak area.The average recovery rate was 98.70% , RSD 1.60%(n=6).Conclusion This method can be used to determine the polydatin content in Polygonum Cillinerve(Nakai)Ohwl for controlling the quality of the medicinal material.
Polygonum Cillinerve(Nakai)Ohwl;HPLC;polydatin;content
R284.1;R282.71
A
1006-4931(2011)10-0029-02
2010-07-19;
2011-01-25)
猜你喜欢
今日农业(2022年2期)2022-11-16
昆明医科大学学报(2021年8期)2021-08-13
今日农业(2021年6期)2021-06-09
特种经济动植物(2021年4期)2021-04-19
中成药(2018年7期)2018-08-04
中成药(2018年6期)2018-07-11
中成药(2018年3期)2018-05-07
中成药(2018年1期)2018-02-02
中成药(2017年8期)2017-11-22
中成药(2017年3期)2017-05-17
