
(S)-4-羟基吡咯烷-2-酮的简便合成*
2010-11-27 11:20:04林紫云黄海洪
合成化学 2010年3期
刘 洋, 张 锋, 林紫云, 黄海洪
(北京协和医学院 中国医学科学院 药物研究所,北京 100050)
(S)-4-羟基吡咯烷-2-酮(1)及其衍生物作为环化的γ-氨基丁酸类似物,是合成具有中枢抑制作用γ-氨基-β-羟基丁酸的重要中间体[1],此外,也是作为合成1-β-甲基碳(杂)青霉烯类抗生素药物的关键中间体[2]。鉴于1在药物合成中的重要应用,采用手性起始原料获得1的合成方法研究受到了广泛关注。根据所用的起始原料分类主要包括以下方法:(1)以(S)-苹果酸[3]为起始原料,通过甲酯化、选择性还原、对甲苯磺酰化及在氨水作用下合环制备1,但是还原反应使用了较贵的硼烷二甲硫醚,而且最终产物的纯化采用离子交换树脂,后处理较为繁琐。黄培强等[4]也以(S)-苹果酸为起始原料制备了1,在脱除内酰胺环氮上的苄基保护时,采用空气敏感的丁基锂及萘锂试剂,收率较低。(2)以(S)-4-氯-3-羟基-丁酸乙酯[2]为原料,依次对其进行叠氮化、羟基保护、酯基在低温下采用DIBALH还原成醛,最后合环、脱保护,虽然5步反应总收率达40%,但反应条件较为苛刻。(3)以(S)-3-羟基-γ-丁内酯[5]为起始原料,经羟基保护、苄胺或烯丙胺开环得到酰胺化合物,再合环、脱保护制备1,虽然总收率高达70%,但是关键步骤的反应条件苛刻,尤其是脱内酰胺环氮上的苄基保护基时,需要使用Li/NH3(液),脱烯丙基保护基时需要使用昂贵的三氯化铑。
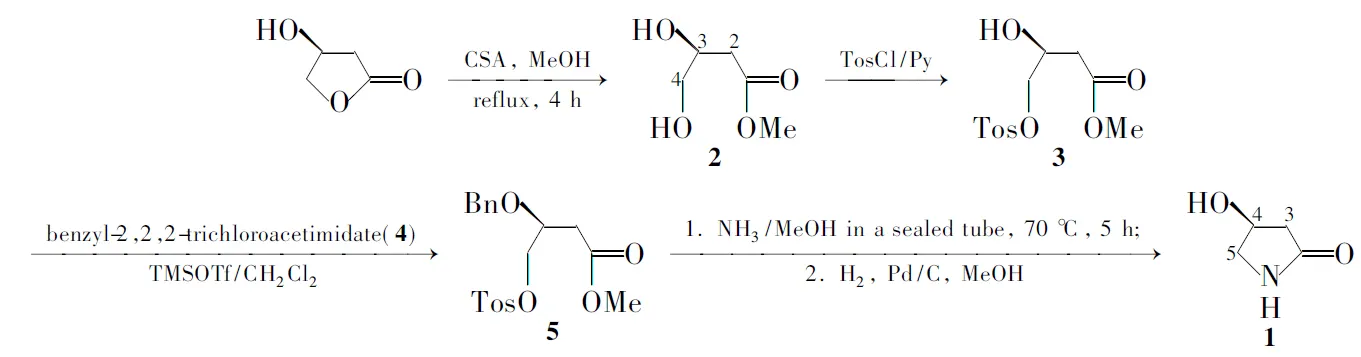
Scheme 1
在文献[2~5]方法的基础上,本文以(S)-3-羟基-γ-丁内酯为起始原料,对开环后所得的3,4-二羟基丁酸甲酯(2)的伯羟基进行选择性对甲苯磺酰化[3],然后对仲羟基进行苄基保护,得到关键中间体4-对甲苯磺酰氧基-3-苄氧基丁酸甲酯(5); 5在氨甲醇的作用下合环、经Pd/C催化氢化脱除苄基保护合成了1(Scheme 1),总收率26%,其结构经1H NMR和HR-MS确认,比旋光值与文献值相符。
1 实验部分
1.1 仪器与试剂
Yanaco MP-500型熔点仪(温度计未校正);Perkin Elmer Model 341LC型旋光仪;MERCURY-300型核磁共振仪(CDCl3为溶剂,TMS为内标);Agilent LC/MSD TOF型液相色谱质谱联用仪(LC-MS)。
所用试剂均为分析纯或化学纯。
1.2 合成
(1) 2的合成
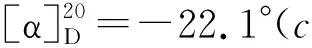
(2) 3的合成[3]
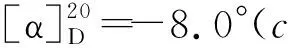
(3) 三氯乙酰亚胺苄酯(4)的合成[8]
在三口瓶中加入苄醇5.40 g(50 mmol)的二氯甲烷(50 mL)溶液,搅拌下于-15 ℃加入50%氢氧化钾溶液50 mL和四丁基硫酸氢铵8 mg,反应10 min;滴加三氯乙腈8.67 g(60 mmol)(维持-15 ℃~-10 ℃),滴毕,反应30 min;于室温反应30 min。分液,水层用二氯甲烷洗涤,合并有机层,用无水硫酸钠干燥,减压浓缩得淡黄色油状液体4 12.17g,收率96.39%,未经进一步纯化,直接用于下步反应。
(4) 5的合成
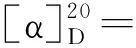
(5)1的合成
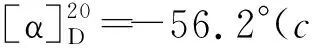
2 结果与讨论
与文献[3]方法相比,本方法具有如下优点:(1)采用(S)-3-羟基-γ-丁内酯为起始原料,经D-樟脑-10-磺酸开环得到二醇化合物2,避免了采用以(S)-苹果酸为起始原料使用硼烷二甲硫醚还原得到二醇的方法;(2)对仲羟基进行苄基保护后,使得合环反应无需在含水介质中进行,且合环产物无需分离纯化直接经Pd/C催化氢化脱除苄基保护,即可得到纯度高的目标化合物,从而避免了采用离子交换树脂的纯化方式,使后处理操作更为简便。
本文采用价廉易得的(S)-3-羟基-γ-丁内酯为起始原料,经4步反应以26%的总收率制备1。合成步骤较少,合成过程中所使用的试剂均较为便宜,反应条件温和,反应操作及产品纯化处理较为简单,是一种较适合于放大量制备的简便方法。
[1] R.Pellegata, I Dosi. (-)-β-Pinene as chiral promoter.Stereospecific access to (-)-γ-amino-β-(R)-hydroxybutyric acid(GABOB) and (R)-carnitine[J].Tetrahedron,1985,41(23):5607-5613.
[2] Satoshi Kobayashi, Katsuhiro Kobayashi. Trials for the synthesis of (R)-4-mercapto-pyrrolidin-2-one[(R)MPD][J].Synlett,1999,S1:909-912.
[3] Masahiko Seki, Kazuhiko Kondo. A facile synthesis of (S)-4-hydroxypyrrolidin-2-one from (S)-malic acid[J].Synthesis,1999,5:745-747.
[4] Pei Qiang Huang, Xiao Zheng. A new approach to (S)-4-hydroxy-2-pyrrolidinone and its 3-substituted analogues[J].Tetrahedron:Asymmetry,1999,10:3309-3317.
[5] Osamu Kanno, Masao Miyauchi, Isao Kawamoto. Efficient syntheses of (S)-4-hydroxy-2-pyrrolidinone derivatives[J].Heterocycles,2000,53(1):173-181.
[6] Norio Nakamura, Hideki Miyazaki. Synthesis and biological activities of bioisostericO-carba-analogues of platelet activating factor(PAF)[J].Chem Pharm Bull,1984,32(6):2452-2455.
[7] Agnes Pommier, Jean-Marc Pons. The first total synthesis of (-)-lipstatin[J].J Org Chem,1995,60:7334-7339.
[8] Vijay J Patil. A Simple access to trichloroacetimidates[J].Tetrahedron Letters,1996,37(9):1481-1484.
[9] Xue-song Chen, Yu-lin Wu. Structure determination and synthesis of a new cerebroside isolated from the traditional Chinese medicine Typhonium giganteum Engl[J].Tetrahedron Letters,2002,43:3529-3532.
猜你喜欢
现代电力(2022年2期)2022-05-23 12:46:08
中国饲料(2021年17期)2021-11-02 08:15:10
当代水产(2021年2期)2021-03-29 02:57:48
传染病信息(2021年6期)2021-02-12 01:52:14
科学与财富(2017年16期)2017-06-13 18:52:19
电子测试(2017年23期)2017-04-04 05:07:20
四川电力技术(2016年3期)2016-08-25 08:11:57
合成化学(2015年10期)2016-01-17 08:55:42
合成化学(2015年1期)2016-01-17 08:53:55
烟草科技(2015年8期)2015-12-20 08:27:06
