
N-取代苄基-N'-苯甲酰氧乙基哌嗪的合成
2010-10-26 11:08韩生华孟双明温雪山陈建新
山西农业大学学报(自然科学版) 2010年1期
韩生华,孟双明,温雪山,陈建新
(1.山西大同大学 化学与化工学院;山西 大同037009;2.山西师范大学化学与材料学院,山西 临汾041004)
近年来,哌嗪类化合物作为药物研发越来越受到人们的广泛关注。根据哌嗪的结构,人们对哌嗪的1,4位取代基进行了大量的研究,发现它具有精神抑制药、抗菌、镇咳和治疗心血管等性质[1~4],从而研制出 Buspirone、Gepirone、Itraconazole、levodropropizine、Ranolazine 等药物[5~7]。根据药学上的拼合原理,我们以哌嗪为母体,在其1,4位上分别接上烷基和苄基,设计合成了六个取代基哌嗪类衍生物(3a-f),其合成路线见图1,期望筛选出具有活性的药物。
该化合物的合成步骤如下:
1 实验部分
1.1 仪器和试剂
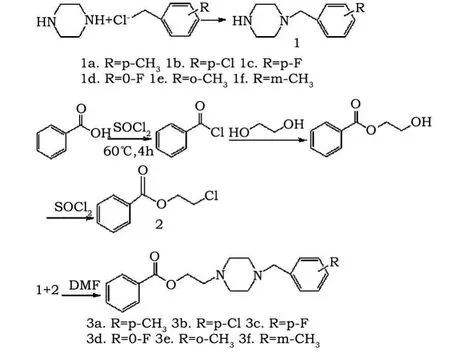
图1 化合物3a-f的合成路线Fig.1 Synthetic route of compounds 3a-f
X-4数字显微熔点仪(温度计未校正);PE-1710型红外光谱仪(KBr压片或液膜);DPX-400型超导核磁共振仪(氘代氯仿为溶剂,TMS为内标);CE-1106型元素分析仪;快速色谱柱或制备薄层色谱,青岛海洋化工厂生产的GF254型硅胶板;ZF-20D暗箱式紫外分析仪;哌嗪为工业品,其余试剂均为分析纯。
1.2 1-(取代苄基)哌嗪的合成
化合物(1a)的合成。将192 g(2.25mol)哌嗪,1500 mL新制的蒸馏水和900 mL异丙醇加入反应瓶中,搅拌下冷却至0~5℃,在此温度下滴加4-甲基氯苄105 g(0.75 mol,99 mL)与600 mL异丙醇的混合溶液,约5~6 h滴毕,室温下搅拌过夜,常压86℃蒸出异丙醇,用三氯甲烷萃取3次,无水硫酸钠干燥,次日蒸除溶剂,得白色固体116 g,产率:81.8%。1H NMR(CDCl3)δ(mg/kg):7.15 ~7.19(m,4H,Ar-H),3.38(s,2H,CH2),2.64(s,4H,pip),2.23(s,4H,pip),2.27(s,3H,CH3),3.18(s,1H,NH);IR(KBr,cm-1):3415,3240,1720,1605,1325,1250;MS(E SI)m/z:200.2742(M+H)+;元素分析(C12 H 18 N2),计算值(%):C 75.74,H 9.53,N 14.72;实测值(%):C 74.89,H 9.62,N 14.83。
化合物(1b)的合成。将192 g(2.25 mol)哌嗪,1500 mL新制的蒸馏水和900 mL异丙醇加入反应瓶中,搅拌下冷却至0~5℃,在此温度下滴加4-氯苄基氯121 g(0.75 mol,95 mL)与600 mL异丙醇的混合溶液,约5~6 h滴毕,室温下搅拌过夜,常压86℃蒸出异丙醇,用三氯甲烷萃取3次,无水硫酸钠干燥,次日蒸除溶剂,得无色液体116 g,产率:73.5%。1H NMR(CDCl3)δ(mg/kg):7.25 ~7.26(m,4H,Ar-H),3.44(s,2H,CH2),2.87(s,4H,pip),2.38(s,4H,pip),1.58(s,1H,NH);IR(KBr,cm-1):3427,3186,1719,1620,1407,1384;MS(ESI)m/z:211.7126(M+H)+;元素分析(C11H 15 N2F),计算值(%):C 62.70,H 7.18,N 13.30;实测值(%):C 62.86,H 7.25,N 13.27。
化合物(1c)的合成。将192 g(2.25 mol)哌嗪,1500 mL新制的蒸馏水和900 mL异丙醇加入反应瓶中,搅拌下冷却至0~5℃,在此温度下滴加4-氟苄基氯108 g(0.75 mol,90 mL)与600 mL异丙醇的混合溶液,约5~6 h滴毕,室温下搅拌过夜,常压86℃蒸出异丙醇,用三氯甲烷萃取3次,无水硫酸钠干燥,次日蒸除溶剂,得白色固体117 g,产率:80.5%。1H NMR(CDCl3)δ(mg/kg):6.99 ~7.28(m,4H,Ar-H),3.45(s,2H,CH2),2.87(s,4H,pip),2.39(s,4H,pip),1.59(s,1H,NH);IR(KBr,cm-1):3426,3142,1719,1638,1507,1436;MS(ESI)m/z:195.2569(M+H)+;元素分析(C11 H15N2F),计算值(%):C 68.01,H 7.78,N 14.42;实测值(%):C 67.86,H 7.65,N 14.26。
化合物(1d)的合成。将192 g(2.25 mol)哌嗪,1500 mL新制的蒸馏水和900 mL异丙醇加入反应瓶中,搅拌下冷却至0~5℃,在此温度下滴加2-氟苄基氯108 g(0.75 mol,91 mL)与600 mL异丙醇的混合溶液,约5~6 h滴毕,室温下搅拌过夜,常压86℃蒸出异丙醇,用三氯甲烷萃取3次,无水硫酸钠干燥,次日蒸除溶剂,得无色液体109 g,产率:75.01%。1H NMR(CDCl3)δ(mg/kg):6.85 ~7.18(m,4H,Ar-H),3.45(s,2H,CH 2),2.87(s,4H,pip),2.38(s,4H,pip),1.62(s,1H,NH);IR(KBr,cm-1):3427,2892,1696,1672,1543,1285;MS(ESI)m/z:195.2569(M+H)+;元素分析(C11H 15 N2F),计算值(%):C 68.01,H 7.78,N 14.42;实测值(%):C 68.13,H 7.62,N 14.39。
化合物(1e)的合成。将192 g(2.25 mol)哌嗪,1500 mL新制的蒸馏水和900 mL异丙醇加入反应瓶中,搅拌下冷却至0~5℃,在此温度下滴加2-甲基氯苄105 g(0.75 mol,97 mL)与600 mL异丙醇的混合溶液,约5~6 h滴毕,室温下搅拌过夜,常压86℃蒸出异丙醇,用三氯甲烷萃取3次,无水硫酸钠干燥,次日蒸除溶剂,得白色固体112 g,产率:78.97%。1H NMR(CDCl3)δ(mg/kg):7.08 ~7.22(m,4H,Ar-H),3.38(s,2H,CH2),2.64(s,4H,pip),2.21(s,4H,pip),2.25(s,3H,CH 3),3.17(s,1H,NH);IR(KBr,cm-1):3421,2865,1734,1682,1462,1275;MS(ESI)m/z:200.2864(M+H)+;元素分析(C12 H 18 N 2)计算值(%):C 75.74,H 9.53,N 14.72;实测值(%):C 74.96,H 9.49,N 14.34。
化合物(1f)的合成。将192 g(2.25 mol)哌嗪,1500 mL新制的蒸馏水和900 mL异丙醇加入反应瓶中,搅拌下冷却至0~5℃,在此温度下滴加3-甲基氯苄105 g(0.75 mol,96 mL)与600 mL异丙醇的混合溶液,约5~6 h滴毕,室温下搅拌过夜,常压86℃蒸出异丙醇,用三氯甲烷萃取三次,无水硫酸钠干燥,次日蒸除溶剂,得无色液体108 g,产率:76.15%。1H NMR(CDCl3)δ(mg/kg):6.99 ~7.19(m,4H,Ar-H),3.38(s,2H,CH 2),2.64(s,4H,pip),2.21(s,4H,pip),2.24(s,3H,CH 3),3.16(s,1H,NH);IR(KBr,cm-1):3420,2922,1733,1617,1351,1282;MS(E SI)m/z:200.2934(M+H)+;元素分析(C12 H18 N2)计算值(%):C 75.74,H 9.53,N 14.72;实测值(%):C 74.62,H 9.38,N 14.16。
1.3 苯甲酸-2-氯乙酯的合成[8]
将 36.3 g(0.3 mol)苯甲酸与45 mL(0.6 mol)二氯亚砜加热搅拌回流4 h,后转入蒸馏,蒸出100℃下液体,再进行减压蒸馏收集42℃/0.9~1 MPa的馏分,得苯甲酰氯(无色液体)38 g,产率90.90%;将制的苯甲酰氯35 mL(0.25 mol)与150 mL三氯甲烷混合后,温度控制在10℃下,缓慢滴加到42 mL(0.75 mol)乙二醇与23 mL(0.26 mol)吡啶的75 mL三氯甲烷溶液中,搅拌过夜,处理得无色液体35.5g,产率85.5%;后与氯化亚砜制的化合物 2。1H NMR(CDCl3)δ(mg/kg):7.444~8.102(m,5H,Ar-H),4.588(t,2H,CH2),3.834(t,2H,CH 2);IR(KBr,cm-1):3025,1811,1719,1626,1438,1424,1328,1125;MS(ESI)m/z:185.6254(M+H)+;元素分析(C9H 9O2Cl),计算值(%):C 58.55,H 4.91;实测值(%):C 58.43 H 4.82。
1.4 N-取代苄基-N'-苯甲酰氧乙基哌嗪的合成
化合物(3a)的合成是将0.95 g(0.005mol)1a、0.92g(0.005mol)2和 20 mL DMF,加入 1.38 g K2 CO3和少许KI。加热至140℃,搅拌回流4 h,冷却过滤,滤液蒸出DMF,得白色固体1.54 g,产率91.1%。用无水乙醇重结晶得3a 1.26g,产率 74.5%。1HNMR(CDCl3)δ(mg/kg):7.101~8.030(m,9H,Ar-H),4.426(s,2H,CH2),3.461(s,2H,CH 2),2.787(s,2H,CH 2),2.601(s,4H,pip),2.325~2.485(s,4H,pip),1.813(s,3H,CH3);IR(KBr,cm-1):3425,3140,2947,2811,1719,1603,1458,1405,1284,1130,1009;MS(ESI)m/z:339.4562(M+H)+;元素分析(C21 H 26 N2O2),计算值(%):C 74.52,H 7.74,N 8.28;实测值(%):C 74.69,H 7.63,N 8.48。
化合物(3b)的合成是将1.05 g(0.005 mol)1b、0.92 g(0.005 mol)2 和 20 mL DMF,加入1.38g K 2CO3和少许KI。加热至140℃,搅拌回流4h,冷却过滤,滤液蒸出DMF得白色固体1.68g,产率93.8%。用无水乙醇重结晶得3b 1.35g,产率 75.3%。1H NMR(CDCl3)δ(mg/kg):7.281~8.057(m,9H,Ar-H),4.471(s,2H,CH 2),3.481(s,2H,CH2),2.795~2.834(s,2H,CH 2),2.628(s,4H,pip),2.189~2.493(s,4H,pip);IR(KBr,cm-1):3430,3139,2947,2816,1719,1580,1400,1284,1125;MS(ESI)m/z:359.8724(M+H)+;元素分析(C20 H 23 N2O2Cl),计算值(%):C 70.06,H 6.76,N 8.17;实测值(%):C 70.29,H 6.85,N 8.24。
化合物(3c)的合成是将0.97 g(0.005 mol)1c、0.92 g(0.005mol)2 和20 mL DMF,加入1.38 g K 2CO3和少许KI。加热至140℃,搅拌回流4 h,冷却过滤,滤液蒸出DMF得白色固体1.56g,产率91.4%。用无水乙醇重结晶得3c 1.29g,产率75.6%。1H NMR(CDCl3)δ(mg/kg):6.974 ~8.050(m,9H,Ar-H),4.465(s,2H,CH 2),3.479(s,2H,CH 2),2.789-2.829(s,2H,CH 2),2.629(s,4H,pip),2.185-2.494(s,4H,pip);IR(KBr,cm-1):3425,3135,1719,1603,1516,1405,1284,1125;MS(ESI)m/z:343.4126(M+H)+;元素分析(C20 H 23 N 2O22F),计算值(%):C 70.15,H 6.77,N 8.18;实测值(%):C 70.34,H 6.86,N 8.29。
化合物(3d)的合成是将0.97 g(0.005mol)1d、0.92 g(0.005mol)2 和 20mL DMF,加 入1.38g K 2CO3和少许KI。加热至140℃,搅拌回流4 h,冷却过滤,滤液蒸出 DMF得白色固体1.45g,产率 84.8%。用无水乙醇重结晶得 3d 1.12g,产 率 65.5%。1H NMR(CDCl-3)δ(mg/kg):6.984~8.029(m,9H,Ar-H),4.418~4.457(s,2H,CH 2),3.595(s,2H,CH 2),2.764~2.804(s,2H,CH2),2.622(s,4H,pip),2.553(s,4H,pip);IR(KBr,cm-1):3437,2857,2635,1658,1523,1459,1276,1148;MS(ESI)m/z:343.4116(M+H)+;元素分析(C20H23N2O2F),计算值(%):C 70.15,H 6.77,N 8.18;实测值(%):C 70.26,H 6.68,N 8.32。
化合物(3e)的合成是将0.95 g(0.005 mol)1e、0.92 g(0.005 mol)2 和 20mL DMF,加入1.38g K 2CO3和少许KI。加热至140℃,搅拌回流4 h,冷却过滤,滤液蒸出 DMF得白色固体1.61g,产率 95.4%。用无水乙醇重结晶得3e 1.29g,产率 76.4%。1H NMR(CDCl3)δ(mg/kg):7.196~8.008(m,9H,Ar-H),4.457(s,2H,CH2),3.084~3.106(s,2H,CH2),2.753(s,2H,CH 2),2.329~2.543(s,4H,pip),2.373(s,4H,pip)1.386~1.434(s,3H,CH 3);IR(KBr,cm-1):3421,2853,2649,2261,1716,1692,1453,1295,1193,1024;MS(ESI)m/z:339.4327(M+H)+;元素分析(C21H26N2O2),计算值(%):C 74.52,H 7.74,N 8.28;实测值(%):C 74.48,H 7.50,N 8.37。
化合物(3f)的合成是将0.95 g(0.005 mol)1f、0.92g(0.005mol)2 和 20mL DMF,加入1.38g K2CO3和少许KI。加热至140℃,搅拌回流4 h,冷却过滤,滤液蒸出 DMF得白色固体1.53g,产率 90.3%。用无水乙醇重结晶得 3f 1.19g,产率 70.2%。1H NMR(CDCl3)δ(mg/kg):6.184~8.006(m,9H,Ar-H),4.495(s,2H,CH2),3.697(s,2H,CH2),2.108(s,2H,CH 2),2.980~3.085(s,4H,pip),2.357(s,4H,pip)1.385~1.434(s,3H,CH 3);IR(KBr,cm-1):3420,3135,2922,2603,1729,1608,1410,1279,1115;MS(ESI)m/z:339.4385(M+H)+;元素分析(C21 H 26 N2O2),计算值(%):C 74.52,H 7.74,N 8.28;实测值(%):C 74.87,H 7.52,N 8.34。
2 结果与讨论
由于哌嗪具有对称性结构,1,4位的N具有相同的活性,在制备哌嗪1,4位不同取代基的衍生物时,首先要考虑的是如何能有效的生成单取代哌嗪产物,控制反应条件使反应第一步不发生或少发生双取代反应[9]。实验结果表明:在不保护胺基的条件下,直接以哌嗪为原料,使用过量3倍的哌嗪,避免引入其它无机碱。同时我们采用了极稀的混合溶液作为溶剂,保持常温下搅拌状态,选择不同混合溶剂(水和异丙醇)的比例,发现比例为1∶1时,能达到较高的产率(81.8%)。
在3a的制备过程中,我们选择了不同的溶剂,实验结果见表1,当用DMF作溶剂时,保持回流状态下,TLC监控反应,4 h后反应完全,产率可达91.1%,其它化合物按照3a条件进行。
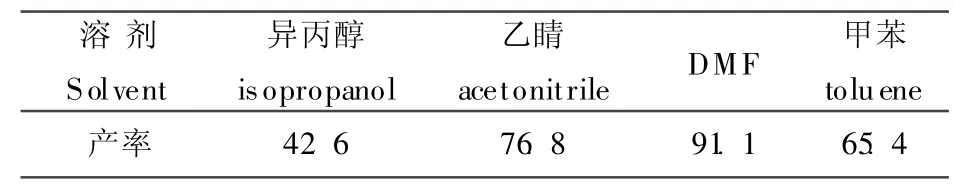
表1 溶剂的影响Table 1 Effect on the solvent
3 结论
由以上分析我们得出了合成哌嗪类衍生物的最佳条件:在合成1-(取代苄基)哌嗪中,哌嗪与取代氯苄摩尔数之比3∶1,常温下搅拌过夜,产率很高。在合成N-取代苄基-N'-苯甲酰氧乙基哌嗪中,用DMF做溶剂成功制备了六个哌嗪类衍生物。
[1]杨键,向钰.镇咳药左旋羟苯哌嗪的合成[J].药物研究,2000,9(3),25.
[2]徐叔云,魏 伟.临床药理学[M].北京.人民卫生出版社,2006:213-214.
[3]王海琴,朱航昌,陈国华.新型抗心绞痛药物雷诺嗪的合成工艺改进[J].Pharmac Clin Res,2007,15(4),282-284.
[4]Ohtaka H,Kanazawa K,Ito K.benzylpiperazine Derivatives.Synthese and vasodilating activities of 1-benzyl-4-dipheny lmethy l piperazine derivatives[J].Chem Pharm Bull,1987,35(8):3270-3275.
[5]周萍,倪沛洲,王礼琛,等.4-取代的1-[二-(4-氟苯基)甲基]哌嗪化合物的合成及生物活性[J].中国药科大学学报,2002,33(6):473-477.
[6]Odds F C.Resistance of yeasts to azole-derivative antifungals[J].J Antimicrob Chemother,1993,31(4):463-471.
[7]王娟,曹洁.阿米三嗪在呼吸系统疾病中的应用进展[J].国际呼吸杂志,2006,26(11):830-832.
[8]Ford-Moore A H.Benzoy lcholine Iodide and Chloride[J].Organ Synth,1963,30(4):84.
[9]廖新成,荣垂林,武现丽,等.1-[二-(4-氟苯)甲基]-4-酰基取代哌嗪类衍生物的合成[J].化学研究与应用,2004,16(4):496-498.
猜你喜欢
安徽化工(2021年6期)2021-12-02
食品安全导刊(2021年36期)2021-03-14
科学导报(2020年75期)2020-12-21
氯碱工业(2020年6期)2020-03-01
合成化学(2015年10期)2016-01-17
合成化学(2015年10期)2016-01-17
合成化学(2015年10期)2016-01-17
合成化学(2015年1期)2016-01-17
中国洗涤用品工业(2015年9期)2015-02-28
应用化工(2014年7期)2014-08-09
