
PAN基预氧化纳米纤维毡的微波碳化、微波活化
2010-10-19 06:41周美华
化工进展 2010年4期
余 阳,周美华
(东华大学环境科学与工程学院,上海 201620)
研究开发
PAN基预氧化纳米纤维毡的微波碳化、微波活化
余 阳,周美华
(东华大学环境科学与工程学院,上海 201620)
以聚丙烯腈基预氧化纳米纤维毡为原料,在氮气保护下微波碳化制备纳米碳纤维,并以水蒸气为活化剂,通过微波活化正交实验制备活性纳米碳纤维;纳米碳纤维的形态和结构变化通过场发射电镜、红外光谱仪、X衍射仪等仪器进行表征;通过对活性纳米碳纤维的比表面积、孔容、孔径分布、苯酚吸附值的测定,了解正交实验各因素对活性纳米碳纤维活化收率和吸附性能影响的强弱程度。研究结果表明:本研究实验条件下可制备出孔径分布以微孔为主,比表面积达1107.4 m2/g,苯酚吸附值达428.1 mg/g的活性纳米碳纤维。
微波碳化;微波活化;活性纳米碳纤维;苯酚吸附值;孔径分布
活性碳纤维是一种广泛使用的高效吸附材料,它具有微孔分布均匀,透气性好,比表面积大,吸附速度快,易脱附,强度高、寿命长等优点。适用于脱色脱臭,溶剂回收、空气和水质过滤净化、防毒口罩等领域。
静电纺纳米纤维直径在200~800 nm之间,同碳纤维比较,纤维直径更细,因此静电纺纳米纤维经过预氧化、炭化、活化制备出的活性纳米碳纤维将能有更大的比表面积和吸附速率,因而有更大的应用潜力。
传统的碳化、活化方式中的加热模式是通过热传导加热,热量由表面传到内部。这种传热方式达到热平衡需要较长的时间,而且加热环境开放,这样就造成了热量的大量散失。同目前所提倡的节能减排生产工艺要求不适应。
微波是一种电磁波。微波作用到某些物质上时,能产生极化现象。极化现象可分为电子极化、原子极化、界面极化及偶极转向极化等多种类型,其中偶极转向极化对物质的加热起主要作用。极性电介质的分子在外加微波电磁场的作用下,原来杂乱无章的极性分子随之快速改变方向,由于电磁场的变化速度高,高速的轮摆运动,使分子间摩擦产生热能,由此使得物质本身加热升温[1]。
微波加热效果与热传导和对流方式不同,其能量转变为加热物质分子的能量所需的时间近似即时,可达到快速加热的目的,同时也避免了长时间加热所造成的热散失,是一种节能的加热方式。因此,碳化、活化采用微波加热,可以大大缩短工艺流程的生产时间和成本。
1 实 验
1.1 原料、实验装置和表征方法
1.1.1 原料
自制聚丙烯腈(PAN)基预氧化纳米纤维毡。
在山东大学徐忠波等沉淀共聚合制备聚丙烯腈的研究成果[2]基础上,采用混合溶剂法制备黏均分子量180000的聚丙烯腈;将制备出的聚丙烯腈配成纺丝液,通过高压静电纺丝装置制备出纳米纤维毡;纳米纤维毡悬挂状态放入高温鼓风干燥箱中,空气氛围热处理制备出聚丙烯腈基预氧化纳米纤维毡。预氧化纳米纤维毡整体质地柔软。
1.1.2 实验装置
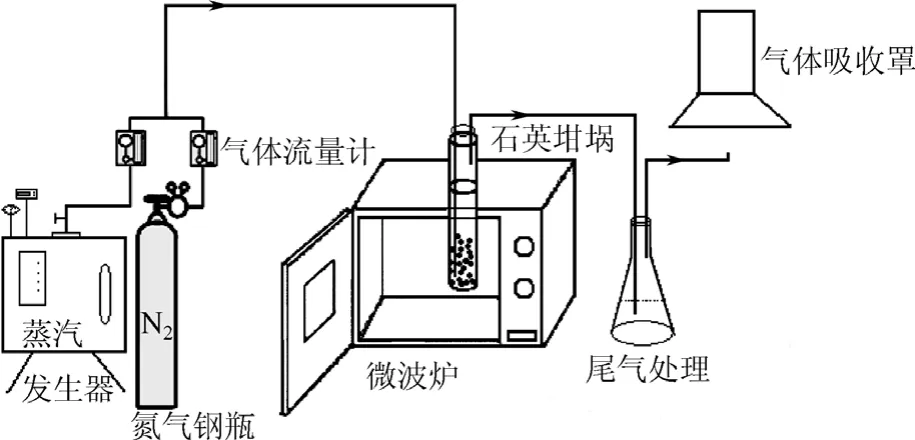
图1 微波活化装置示意图
微波活化装置见图1,主要由高纯氮气钢瓶、水蒸气发生器(江心牌3 kW全自动变频式蒸汽发生机,浙江台州)、石英坩埚、微波加热装置(700 W,上海博奥微波能设备有限公司)和尾气处理装置等部分构成。当水蒸气发生装置关闭时,装置作为微波碳化实验装置使用。
1.1.3 表征方法
采用场发射扫描电镜(S-4800,HITACHI 公司)对微波碳化纳米纤维毡形态进行表征;采用X-ray衍射仪(D/Max-2550 PC,RIGAKU 公司 )和傅里叶变换红外-拉曼光谱仪(NEXUS-670,NICOLET 公司)对微波碳化前后纳米纤维毡结构变化进行表征;采用比表面测试仪(JW-K,北京精微高博)和苯酚吸附值(GB/T7702.8—87)对活性纳米碳纤维的吸附性能进行表征。采用美国康塔仪器公司的全自动比表面和孔径分布分析仪(Autosorb-1,Quantachrome)对活性纳米碳纤维孔容和孔径分布进行表征。
1.2 微波碳化
把1.5 g(精确至0.0001 g)预氧化纳米纤维毡放入图1石英坩埚内,接好装置,通入氮气,开启微波炉加热。微波频率2450 MHz,微波功率700 W,氮气流量为1.5 L/min,碳化时间4 min。
1.3 微波活化正交实验
根据PAN纳米纤维毡微波活化的特点[3],取活化效果影响较大的微波功率、活化时间和水蒸气流量作为微波活化实验的3因素,每因素选取3水平,正交实验因素水平见表1。
用L9(34)正交表[4](见表2)设计实验,其中,第4个因素项下全部为0,以a、b、c分别表示水平1、水平2、水平3。
实验步骤:按正交实验设计,把0.8 g(精确至0.0001g)纳米碳纤维毡放入石英坩埚里,接好装置,通入氮气,氮气流量为1.0 L/min,通水蒸气,开启微波炉加热活化纳米碳纤维毡(微波频率2450 MHz),根据重量法求各实验样品活化收率,并对样品进行比表面积和苯酚吸附值的测定。

表1 微波活化正交实验因素水平表

表2 L9(34)正交实验表
2 结果与讨论
2.1 微波碳化后纤维毡形态和结构变化
图2为微波碳化制备的纳米碳纤维场发射电镜图,由图2可看出,微波碳化制备出的碳纳米纤维其表面和内部结构和形态均一,没有皮芯结构。而且,碳化后的纤维毡其整体质地依然柔软,未发生的碳化变脆情况。

图2 纳米碳纤维纤维结构形态场发射电镜图
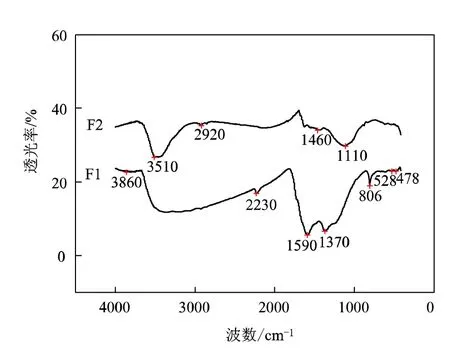
图3 预氧化毡和纳米碳纤维毡的红外光谱图
图3中F1和F2分别为预氧化毡和纳米碳纤维毡的FTIR图谱。从预氧化毡红外曲线F1可以看到:1370 cm-1处有—CH3对称变形振动吸收峰;2230 cm-1处有—C≡N伸缩振动吸收峰;1590 cm-1处有—C=C—C=N—共扼结构吸收峰[5];1370 cm-1处有甲基面内弯曲振动吸收峰;806 cm-1处有C=C—H面外弯曲振动吸收峰。经过微波碳化处理后,纳米碳纤维毡红外曲线F2发生了变化。CH3对称变形振动吸收峰、—C≡N吸收峰、甲基面内弯曲振动吸收峰、C=C—H面外弯曲振动吸收峰消失;—C=C—C=N—共扼结构特征峰显著减弱;但3510 cm-1处为羟基的伸缩振动吸收峰并未消失,并在1110 cm-1处新产生了较稳定的含氧基团特征吸收峰,指示了醚基的存在,其原因可解释为:随着碳化作用,纤维内部的结构发生重排,不稳定基团结构消失,并生成有稳定结构的碳化纤维材料,但由于碳化作用并未完全,仍有羟基和较稳定的醚基含氧基团存在于碳化纤维中。Yu Wang等[6]曾研究了以聚丙烯腈基静电纺纳米纤维毡为原料,通过传统电加热方法碳化制备碳纳米纤维,其结果表明:随碳化温度的升高,碳纳米纤维的石墨化程度增高,在较高温度可以清晰看到石墨的指纹结构。并且,在碳化温度分别为873 K、1073 K、1273 K、1473 K条件下,碳纳米纤维毡的红外光谱图在1580 cm-1和1360 cm-1比处均有明显的吸收峰。由此可知:微波碳化同传统电加热比较,其对碳纳米纤维结构的影响是不同的。

图4 预氧化毡和纳米碳纤维毡的X衍射图
图4中X1和X2分别为预氧化纳米纤维毡和纳米碳纤维毡的X衍射图谱,从图4可以看出,两个衍射峰均较宽,反映出了乱层堆叠的紊乱状态。
X1中2θ=25.46°出现的模糊衍射峰,可以认为是乱层结构中微晶(002)晶面衍射峰[7],是纤维内部形成的环结构逐步积累的结果,表明纤维结构的芳构化。而在X2中,衍射峰稍微发生了偏移,在2θ=24.16°附近出现衍射峰,该峰与文献报道的过渡态碳的X衍射图谱的特征峰基本一致[8],证明已经生成碳结构材料。
2.2 活性纳米碳纤维吸附性能及其孔结构
微波活化正交实验制备出的活性纳米碳纤维,其活化收率、比表面积、苯酚吸附值表征结果见表3。
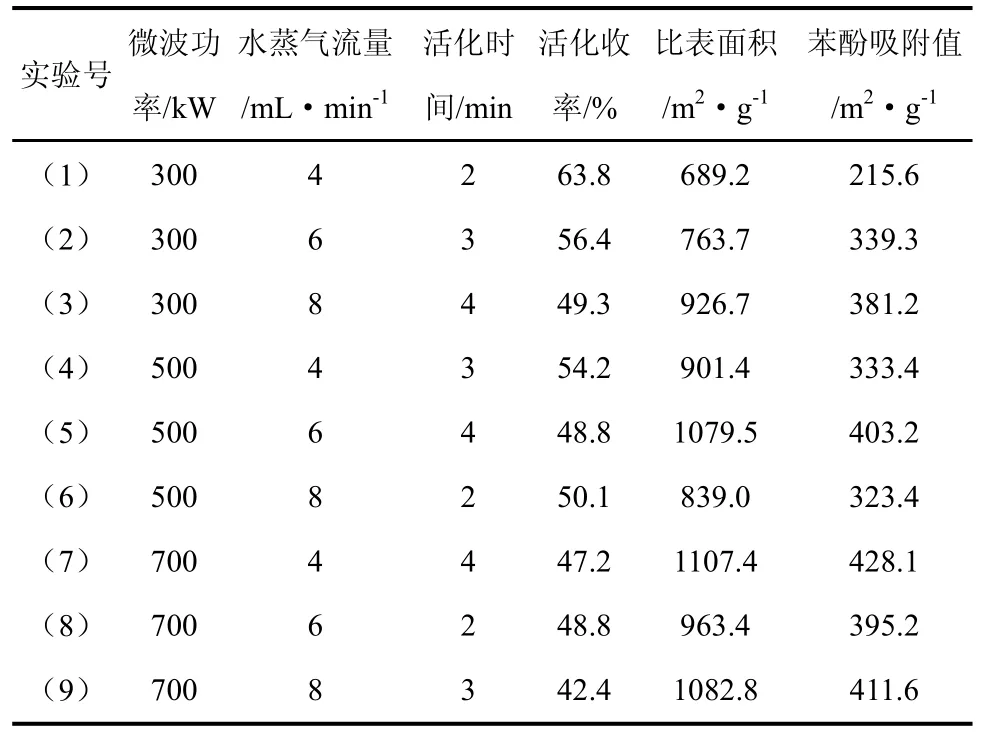
表3 活性纳米碳纤维活化收率、比表面积、苯酚吸附值表征结果
从表3的表征结果可以看出,活化收率和比表面积之间存在相关性,随着活化收率的减少,活性纳米碳纤维的比表面积有增加的趋势,并且苯酚吸附值同比表面积也表现出正相关关系。Martin-Gullon等[9]曾以聚丙烯腈基碳纤维为原料,电加热条件下,活化制备了活性碳纤维,其研究结果表明:用水蒸气作为活化剂制备出的活性碳纤维比表面积最大可达670 m2/g,但活化时间需4 h,活化收率仅14%。吴明铂等以聚丙烯腈预氧纤维为原料,以水蒸气为活化剂,活化温度1073 K,活化时间30 min条件下,只能制备出比表面积达669.12 m2/g的活性碳纤维[10]。由此可见,以水蒸气为活化剂,微波活化制备出的活性纳米碳纤维相对于传统电加热制备出的活性碳纤维,在成本、活化收率和产品性能上均有优势。
为判断所选的3个因素对活性炭收率和吸附性能所产生影响的强弱程度,正交实验表征结果的直观分析见表4。
比较表4各因素的极差可得:对于活化收率,三因素影响强弱程度主次顺序是微波功率>水蒸气流量>活化时间;对于比表面积,三因素影响强弱程度主次顺序是微波功率>活化时间>水蒸气流量;对于苯酚吸附值,三因素影响强弱程度主次顺序是微波功率>活化时间 >水蒸气流量。
采用美国康塔仪器公司的全自动比表面和孔径分布分析仪对表2实验号(7)条件下制备的活性纳米碳纤维样品孔结构进行表征,测得总孔容为0.357 cm3/g,微孔部分总孔容为0.292 cm3/g,其氮吸附等温线和孔径分布图分别见图5、图6。

表4 正交实验表征结果直观分析

图5 77 K条件下N2吸脱附等温线

图6 密度函数理论计算的孔径分布
由图5可知,在77 K条件下,较低的压力范围内,氮吸附很快就接近饱和,并且吸附等温线和脱附等温线几乎完全重合,这证明样品具有很强的吸附势。根据Brunauer,Deming,Deming 和Teller(BDDT)对物理吸附等温线的归纳[11]:该样品的吸附曲线为(Ⅰ)型吸附曲线,其孔径分布以微孔为主。密度函数理论(DFT)表征样品的孔径分布也证实了这点,图6中样品的孔径分布非常集中,绝大部分是2 nm以下的微孔。
实验号(7)条件下,微波活化制备的活性纳米碳纤维场发射电镜见图7。由图可看出,微波活化后,纤维直径略微变小,而且,纤维表面变得粗糙和凹凸不平,这应该是高温活化时水蒸气对碳元素蚀刻和纤维内部气体逸出造成的;并且在放大倍数12万倍的条件下,活性纳米纤维毡表面只能观察到少量中孔,没有观察到大孔结构的存在,这也验证了孔径分布的表征结果。

图7 活性纳米碳纤维纤维结构形态场发射电镜图
2.3 纳米碳纤维活化机理探讨
碳纤维活化反应机理曾被广泛研究过,但因为研究者采用的活化材料不同、材料所含杂质可能存在的催化作用的影响,以及二次反应导致的活化过程过于复杂等原因,使得到目前为止,还未产生能够系统而详细解释活化机理的理论[12]。
需要指出的是:本研究采用的预氧化纳米纤维毡,其在预氧化处理过程中,由于氧原子的进入和非碳原子的溢出,在纤维内部和表面就留有许多“微孔缺陷”;而且预氧化纳米纤维毡碳化制备纳米碳纤维毡过程中,预氧化纤维中所含的小分子物质以及原料本身热分解放出的小分子物质会以气态形式逸出,并在基体中留下“孔隙”[13];这些“微孔缺陷”和“孔隙”在微波碳化过程中又会因为碳骨架的收缩和无定形碳等物质覆盖而闭合形成“闭孔”。另外,碳化过程中可能存在交联反应不充分,热分解不完全,非碳原子未完全脱出情况,这将在碳基体中留下大量“活性点”。
当纳米碳纤维进行微波活化时,伴随着质量损失,碳化过程中形成的“闭孔”被水蒸气活化剂分子打开,成为开孔,随着活化的深入,纳米碳纤维中的大量闭孔将被打开;而在碳化过程中留下的大量“活性点”,也会在活化时伴随着以有机小分子形式脱出,而形成微孔结构。当“闭孔”完全打开,“活性点”消耗完毕后,如果进一步活化,这时生成新微孔较少,活化剂分子将同结晶态的碳发生作用,使结晶态碳消耗而破坏微孔孔壁,使得微孔扩大,或者孔隙合并,形成较大孔隙,进而形成中孔乃至大孔,这一过程同时也会有质量损失。因此可以推断:孔隙的生成量和形态与活化材料的收率应有密切关系。表3活性纳米碳纤维表征结果表明:随着活化收率的减少,活性纳米碳纤维的比表面有增加的趋势。因此可以认为,活化过程就是活化剂刻蚀材料中的无定形碳和部分微晶碳生成孔隙的过程,并且孔隙生成量和形态同活化收率有关。
3 结 论
(1)微波碳化、活化实验表明:微波碳化、活化可以极大的缩短制备活性纳米碳纤维的工艺时间,并可以制备出吸附性能较高的活性纳米碳纤维。
(2)采用美国康塔仪器公司的全自动比表面和孔径分布分析仪表征结果表明:微波活化法可制备出孔径分布以微孔为主,并且微孔孔容在总孔容中占较大比例的活性纳米碳纤维。
(3)活化过程是孔隙生成的过程,而孔隙生成量和形态同活化材料的活化收率存在一定联系。
[1] 叶新强,干占湖,付宣.应用前景广阔的微波处理技术[J].山东环境,1998(1):21-23.
[2] 徐忠波,张旺玺,王成国,等.丙烯腈与衣康酸在混合溶剂中沉淀共聚合[J].化工科技,2002,10(2):1-4.
[3] 季涛,倪朝晖,徐山青,等.静电纺PAN纤维微波活化技术及其吸附性能[J].纺织学报,2006,27(3):16-20.
[4] 袁志发,周静芋.试验设计与分析[M].北京:高等教育出版社,2000:292-296.
[5] Ko T H,Chen C Y. Raman spectroscopic study of the microstructure of carbon films developed from cobalt chloride-modified polyacrylonitrile[J]. J. Appl. Polym. Sci.,1999,71:2219-2225.
[6] Wang Y,Serrano S,Santiago-Avilees J J. Raman characterization of carbon nanofibers prepared using electrospinning[J].Synthetic Metals,2003(138):423-427
[7] Sokot M,Grobelny J,Turska E. Investigation of structural changes of polyacrylonitrile on swelling,wide-angle X-ray scattering study[J]. Polymer,1987,28:843-846.
[8] 杨于兴,汪嘉荣,刘常林.炭纤维的X射线显微结构分析[J].理化检验——物理分册,1989,25(l):46-49.
[9] Martin-Gullon I,Andtews R,Jagtoyen M,et al.,PAN-based activated carbon fiber composites for sulfur dioxide conversion:influence of fiber activation method[J]. Fuel,2001,80:969-977.
[10] 吴明铂,查庆芳,邱介山,等.PAN活性炭纤维表面结构[J].大连理工大学学报,2003(9):591-594.
[11] 陈诵英,孙予军,丁云杰,等. 吸附与催化[M].郑州:河南科学技术出版社,2001:2-4.
[12] 立本英机, 安部郁夫(高尚愚译编).活性炭的应用技术-其维持管理及存在问题[M].南京:东南大学出版社,2002:33-35.
[13] 仲亚娟,孙亚娟,于万秋,等. PAN基炭纤维微孔结构的研究[J]. 吉林师范大学学报:自然科学版,2005(3):16-18.
Microwave carbonization and microwave activation of PAN-based preoxidized nanofiber mat
YU Yang,ZHOU Meihua
(School of Environmental Science & Engineering,DongHua University,Shanghai 201620,China)
Polyacrylonitrile-based preoxidized nanofiber mat was microwave carbonized in nitrogen atmosphere for preparing carbon nanofiber mat,then water steam was taken as activating agent for preparing the activated carbon nanofiber mat. The operation conditions were optimized through orthogonal experiment. Field emission-scanning electron microscope,infrared spectrometer,X-ray diffraction were employed to characterize the morphology and structure of the carbon nanofiber mat. Specific surface area,pore volume,pore size distribution and phenol adsorption value of the obtained carbon nanofiber mat were measured to evaluate the effects of the operation factors in the orthogonal experiment on the yield of activation and adsorption properties of activated carbon nanofiber mat. Results showed that activated carbon nanofiber mat with excellent adsorption properties could be prepared under the optimized operation conditions. The obtained carbon nanofiber mat showed mainly micropores with specific surface area of 1107.4 m2/g and phenol adsorption value of 428.1 mg/g.
microwave carbonization;microwave activation;activated carbon nanofiber mat;phenol adsorption value;pore size distribution
TQ 342.74
A
1000–6613(2010)04–0704–06
2009-09-09;修改稿日期:2009-12-16。
上海市重点学科建设资助项目(B604)。
余阳(1977-),男,博士。电话 021-67792520;E-mail yuyang@dhu.edu.cn。
猜你喜欢
环境卫生工程(2021年4期)2021-10-13
西南石油大学学报(自然科学版)(2018年2期)2018-06-26
雷达学报(2017年1期)2017-05-17
光学精密工程(2016年1期)2016-11-07
中国塑料(2016年4期)2016-06-27
中国塑料(2015年3期)2015-11-27
中国塑料(2015年7期)2015-10-14
水利建设与管理(2015年10期)2015-05-09
重庆建筑(2014年12期)2014-07-24
纯碱工业(2014年6期)2014-03-11
